
Abstract
This study was conducted to develop formulations of hydrocortisone butyrate (HB)-loaded poly(d,l-lactic-co-glycolic acid) nanoparticles (PLGA NP) suspended in thermosensitive gel to improve ocular bioavailability of HB for the treatment of bacterial corneal keratitis. PLGA NP with different surfactants such as polyvinyl alcohol (PVA), pluronic F-108, and chitosan were prepared using oil-in-water (O/W) emulsion evaporation technique. NP were characterized with respect to particle size, entrapment efficiency, polydispersity, drug loading, surface morphology, zeta potential, and crystallinity. In vitro release of HB from NP showed a biphasic release pattern with an initial burst phase followed by a sustained phase. Such burst effect was completely eliminated when nanoparticles were suspended in thermosensitive gels and zero-order release kinetics was observed. In HCEC cell line, chitosan-emulsified NP showed the highest cellular uptake efficiency over PVA- and pluronic-emulsified NP (59.09 ± 6.21%, 55.74 ± 6.26%, and 62.54 ± 3.30%, respectively) after 4 h. However, chitosan-emulsified NP indicated significant cytotoxicity of 200 and 500 μg/mL after 48 h, while PVA- and pluronic-emulsified NP exhibited no significant cytotoxicity. PLGA NP dispersed in thermosensitive gels can be considered as a promising drug delivery system for the treatment of anterior eye diseases.
Similar content being viewed by others


INTRODUCTION
Eye drops are generally formulated as aqueous solutions. Rapid tear turnover, hydrophobic structure of the corneal epithelium, and lachrymal drainage can lower ocular bioavailability to less than 1% [1]. Moreover, a major proportion of a dose (about 90% of the instilled drug) cannot permeate through the cornea. A small fraction of the dose is absorbed into the systemic circulation by the conjunctival and nasal blood vessels, causing secondary effects [2]. Topical corticosteroids are administered by drops, suspension, creams, and ointment for ocular inflammation. A 0.1% hydrocortisone butyrate (HB) ointment displayed no eye irritation in rabbits [3, 4]. However, a conventional ophthalmic formulation of steroids may only achieve short-term relief of inflammation, owing to very limited residence in the precorneal area. Therefore, steroids require frequent administrations, leading to side effects. Lipophilic HB for the treatment of inflammation suffers from poor solubility, low bioavailability, and severe side effects. A few attempts to increasing dissolution and bioavailability of HB have met with limited success [5–8].
Over the last decade, biodegradable nanoparticles (NP) have been developed to regulate drug release, increase accessibility to specific tissue compartments, modulate biodistribution, and enhance bioavailability of poorly soluble drugs [9, 10]. Polymeric NP have been considered as the most promising system for ocular drug delivery [11, 12]. Owing to its excellent biocompatibility, biodegradability, and mechanical strength, poly(d,l-lactide-co-glycolide) (PLGA) has been employed for the delivery of therapeutic agents [13]. It was selected as a matrix to prepare small NP with size <300 nm for ocular administration [14]. The surface properties of these NP such as hydrogen bandies, change, and hydrophobicity were optimized. Such surface modification strengthened adhesion of NP to the cell surface and enhanced uptake, thereby elevating ocular bioavailability and therapeutic efficiency [15]. A longer and closer drug contact with the epithelial ocular surface leads to prolonged corneal penetration [16–18]. In particular, polymeric NP can serve as carriers for the treatment of ophthalmic pathologies, since improved corneal penetration and prolonged therapeutic response may be achieved for specific drugs.
Good tissue tolerability of polymeric NP also depends on the use of well-tolerated surfactants and polymeric stabilizers. Various stabilizers have been added to polymeric NP including ionic, nonionic, and amphoteric surfactants [19, 20]. Polymeric stabilizers are particularly desirable for application due to their deep tissue penetration with low irritation. Owing to their smaller size and zeta potential, pluronic and chitosan may be suitable stabilizers.
Surfactants can be employed to modify surface properties due to their amphiphilic character. Interactions between polymers and surfactants in aqueous solutions have been investigated previously [21]. Polyvinyl alcohol (PVA) polymer is widely used for NP formulations as well as controlled release dosage forms. Nonionic pluronic surfactants are block copolymers of polyoxyethylene sandwiched between polyoxypropylene chains [22]. These surfactants have been broadly utilized for biomedical applications including drug delivery, nonfouling, and tissue engineering applications [23–25]. Amphiphilic polymers due to hydrophobic component can interact with hydrophobic polymer PLGA (75:25), whereas hydrophilic component imparts hydrophilicity.
In an attempt to supply positive charges to the colloidal systems, positive phospholipids and other cationic surfactants have been used in the preparation and stabilization of submicron emulsions [26, 27]. Cationic surfactants will facilitate interactions with negative biological membranes, thus enhancing the capacity for transporting and transferring cargo [28]. Calvo et al. applied chitosan to stabilize the oil/water interface [29, 30]. Chitosan, a linear water-soluble polymer, is prepared by partial alkaline deacetylation of chitin [31]. As a natural positively charged polysaccharide (pKa of 6.3–7), chitosan presents good solubility under acidic conditions due to the protonation of amino group. Chitosan was chosen to coat colloidal systems due to its cationic property, mucoadhesivity, and biocompatibility [32]. The association between a polyelectrolyte and oppositely charged surfactants is of special interest and generally accepted as an ion-exchange process, where electrostatic attraction is strengthened by compliant accumulation of the bound surfactant molecules.
Because of their ultrafine particle size and biodegradable nature, PLGA NP can sustain drug release, and as a result, these colloidal carriers have great potential as controlled release ocular drug delivery systems. However, their utility in achieving the sustained action desired of ocular drug formulation has been questioned since the carriers are drained from the precorneal area rapidly. Therefore, a sustained delivery of HB to the eye may possibly be realized if this colloidal carrier system of HB is incorporated into the PLGA-polyethylene glycol (PEG)-PLGA gel formulation.
The objective of the present study was to develop HB-loaded biodegradable NP incorporated into a PLGA-PEG-PLGA-based gel delivery system which will provide sustained release formulation, prolong the precorneal residence time of drugs, and replace the conventional therapy of eye drop administration. Previously, an emphasis was given to optimization of parameters involved in the emulsification process in favor of the particle size and drug encapsulation efficiency [33]. In this research, we continued our earlier work to develop several biodegradable NP with different surfactants and to investigate the advantages of PF- or chitosan- emulsified PLGA NP over those emulsified by the traditional chemical emulsifier PVA. The HB-loaded PLGA NP were characterized by laser light scattering (LLS) for size distribution, field-emission scanning electron microscopy for surface morphology, and zetasizer for surface charge. High-performance liquid chromatography (HPLC) was selected for drug encapsulation efficiency and in vitro drug release kinetics. The mechanism of encapsulation was studied using differential scanning calorimetry (DSC) and X-ray powder of the optimized formulation. A fluorescent marker, fluorescein isothiocyanate (FITC), was conjugated with polymer PLGA to allow visualization of the cellular uptake. Human corneal epithelial cells (HCEC) were employed to evaluate the cellular uptake of FITC-conjugated drug-loaded PLGA NP and nonfluoresced NP. It was also applied to measure the cytotoxicity of the drug formulation in NP against Taxol®. This work also includes the physical properties of formulation, release mechanism, and the effect of different surfactant types on HB-loaded PLGA NP.
MATERIALS AND METHODS
Materials
HB, PLGA 75:25 with M w 66,000–107,000 Da, and PVA with M w 31,000–50,000 Da were purchased from Sigma-Aldrich Co (St. Louis, MO). Pluronic F108 (PF-108) with average M w 14,600 Da was provided by BASF Corporation (New Jersey). Lactide and glycolide monomers for the synthesis of thermosensitive gel PLGA-PEG-PLGA (PLGA-PEG-PLGA) were donated by Purac America (IL, USA) and PLGA-PEG-PLGA with M w 4759 Da was further synthesized and purified in our laboratory. FITC isomer I for the synthesis of FITC-labeled PLGA 75:25 and chitosan with low molecular weight (75–85% deacetylated) were purchased from Sigma-Aldrich Co (St. Louis, MO). FITC-PLGA was further synthesized in our laboratory [33]. HCEC are obtained as a generous gift from Dr. Araki-Sasaki (Kinki Central Hospital, Japan). Culture flasks (75 cm2 growth area), 12-well Costar@ plates, all buffer components, and solvents were purchased from Fisher Scientific Co. HPLC-grade acetonitrile and dichloromethane were obtained from Fisher Scientific Co (Fairlawn, NJ). Distilled deionized (DDI) water was used in the preparation of formulations, all buffers, and mobile phases. Ultrapure water was prepared using a Milli-Q® water purification system (Millipore Co., Billerica, USA) and filled (0.22 μm) prior to use in mobile phases. All other chemicals in the study were of analytical reagent grade and obtained from Sigma-Aldrich Co.
Methods
HPLC Analytical Method
Samples were analyzed by HPLC system (Waters 600 Pump, Waters, Milford, MA) equipped with a fluorescence detector (HP1100, Hewlett Packard, Waldbronn, Germany). A reversed phase Kinetex 2.6 μm PFP 100 A column (50 × 2.1 mm ID, Torrance, CA, USA) was employed. The mobile phase of acetonitrile and water (38:62, v/v) was kept at the flow rate of 0.6 mL/min at 25°C. The column effluent was detected at 240 nm with a UV-VIS detector at sensitivity 0.0010 AUFS. Sample injection volume was kept at 50 μL. The limit of quantification was found to be 0.4 μg/mL. The retention time (t R) was 7.78 min.
Preparation of NP by W/O Single Emulsion Solvent Evaporation Method
HB-loaded NP were prepared at room temperature by a previously described emulsion solvent evaporation method [33]. Briefly, 12.5 mg/mL of PLGA and 9 mg/mL of HB were dissolved in dichloromethane (5 mL) to form the organic phase. It was then slowly added to an aqueous solution containing the surfactant. For three surfactant solutions, 1.8% PVA solution was prepared by slowly adding PVA into 80°C heating water under stirring for 30 min, then cooled down to room temperature; pluronic F-108 was dissolved in distilled water (pH 6.8) to make a 1% stock solution at 40°C for about 20 min and the solution was stored at room temperature; chitosan was first dissolved in 0.1 M acetic acid (pH 4.7) at 60°C with 5 h stirring to obtain 10% chitosan, then cooled down to room temperature, and further diluted into 1% chitosan solution. An oil/water-type emulsion was formed upon sonication for 5.6 min over an ice bath using a Fisher Scientific 100 ultrasonic probe. Organic solvent was removed by stirring at room temperature for 6 h and then evaporated under a vacuum for 30 min to ensure complete removal of organic solvent. The samples were centrifuged at 1500×g for 10 min to remove undissolved drugs. To remove the unentrapped drug and surfactant, NP suspension was further washed with DDI water followed by centrifugation at 5000×g (22,000 rpm) for 1 h. This procedure was repeated three times. The resulting NP suspension was frozen and lyophilized over 48 h under a freeze-drier system to obtain NP in powder form. FITC-PLGA NP for uptake studies were prepared by blending PLGA and FITC-conjugated PLGA at a 90:10 weight ratio according to double emulsion solvent evaporation method.
Entrapment Efficiency and Drug Loading
Entrapment efficiency and loading of HB in NP were also determined. Freeze-dried NP (1 mg) were dissolved in 2 mL of dichloromethane and vortexed for 1 min to completely destruct the NP. Subsequently, the mixture was dried under inert atmosphere and dissolved in 500 μL of acetonitrile/water (60:40) by vortexing. After centrifugation at 12,000 rpm for 10 min, the supernatant was aspirated to determine the drug amount by HPLC. Entrapment efficiency, drug loading, and yields were calculated by Eqs. 1, 2, and 3, respectively. The measurements were performed in triplicate.
Particle Size and Polydispersity by Dynamic Light Scattering (DLS)
Average particle size and size distribution of NP were measured by dynamic light scattering analyzer (Brookhaven Zeta Plus instrument, Holsville, NY). The dispersion of NP was measured after complete sonication before measurement. It was considered monodispersed when polydispersity index (PI) ≤0.05. Analyzers were performed in triplicate.
Zeta Potential
The surface charge of the NP in water was determined by Nano ZS zetasizer (Malvern Instruments Ltd, Worcestershire, UK) at room temperature. NP were diluted by ultrapure water to obtain 0.005 wt% suspension. All measurements were performed in triplicate.
Surface Morphology Analysis by Scanning Electron Microscope (SEM)
The shape and surface morphology of the NP was determined using a FEG ESEM XL 30 SEM (FEI, Hillsboro, OR, USA). To obtain the specimen, freeze-dried NP were attached to a double-sided tape and spray-coated with gold-palladium at 0.6 kV. NP were viewed under the electron microscope and digital images were recorded.
Differential Scanning Calorimetry
Samples (5 mg) were sealed in aluminum sample pans with an empty pan as reference and then analyzed on a differential scanning calorimeter (DSC-60, Shimadzu Corporation). The samples were heated at the speed of 5°C/min in the range of 30 to 350°C under a nitrogen atmosphere at a flow rate of 20 mL/min.
X-Ray Diffraction (XRD)
XRD patterns were obtained with a high-resolution MiniFlex automated X-ray prototype diffractometer (Rigaku, The Woodlands, TX) based on Ni-filtered Cu Kα radiation under 30 kV and 15 mA. Recordings were made at ambient temperature with the diffraction angle from 2θ = 5° to 40° using steps of 0.05° and a count time of 3 s/step (effectively 1°/min). The diffraction patterns were processed using Jade 8+ (Materials Data, Inc., Livermore, CA).
In Vitro Drug Release from NP Suspended in Buffer and Thermosensitive Gel
HB-loaded NP (5 mg) were dispersed in 1 mL of isotonic phosphate-buffered saline (IPBS, pH 7.4) containing 0.1% (w/v) Tween 80, which can improve the solubility of HB in IPBS. This dispersion was subsequently filled into dialysis bags (M wCO 6275 g/mol). NP (5 mg) were quickly suspended in 1 mL of 23% w/w PLGA-PEG-PLGA aqueous polymer solution (4°C) by vortex in 1 mL tube, and then this suspension was added into dialysis bags by pipettes. The dialysis bags were placed into vials containing IPBS (10 mL) and sodium azide (0.025%, w/v) to prevent microbial growth. Tween 80 (0.02%, w/v) was added into the vial solution to maintain sink condition. The polymer solution inside the bags gelled at 37°C over 30–60 s and entrapped the suspended NP. The vials were kept in a shaker bath at 37 ± 0.5°C with a constant agitation of 60 oscillations/min. At predetermined time intervals, 200 μL of the sample was withdrawn and replaced with equal volumes of fresh buffer. Samples were analyzed by HPLC method. All experiments were carried out in triplicate.
Drug Release Mechanism
Drug release parameters were calculated by the following mathematical models: zero order (Eq. 4), first order (Eq. 5), Higuchi (Eq. 6), and Korsmeyer-Peppas (Eq. 7). Release data were fitted into the model equations in order to identify the release mechanism of drug release from formulations.
Q t , Q 0, and Q ∞ represent the cumulative amount of drug released at time t, initial amount of drug, and total amount of drug in dosage form, respectively. K 0 is the zero-order release rate constant obtained by plotting Q t against time. K 1 represents the first-order release rate constant determined by plotting log(Q t /Q ∞) against time. K h is the Higuchi release rate constant obtained by plotting Q t against the square root of time. K p denotes the release rate constant of the Korsmeyer-Peppas model, and the constant n is the release exponent which is used for characterizing the different release mechanisms. The parameters K p and n can be calculated by plotting log(Q t /Q ∞) against log of time.
Cell Culture
HCEC were cultured in our serum-free medium at 37°C in a humidified environment with 5% CO2. The medium was supplemented with 10% calf serum (nonheat inactivated), 100 U/L of penicillin, 100 U/L of streptomycin, 1.76 g/L lactalbumin enzymatic dehydrolysate, and 1.3 g/L HEPES. It was changed every other day; cells were passaged weekly at a 1/3 split ratio.
NP Uptake in HCEC
For the quantitative study, HCEC were seeded into 12-well coverglass chamber at a density of 250,000 cells/well and grown for 5 days till confluent. Cells were then incubated with NP in Dulbecco’s phosphate-buffered saline (DPBS) at various concentrations. Uptake of NP was carried out for 1 to 4 h at 37°C. After each uptake period, cells were washed three times with DPBS to ensure removal of all free NP. Cells were lysed by adding 1 mL DDI water and then stored in −80°C freezer. Cells were then thawed and scraped into 1 mL DDI. Samples were centrifuged at 1000×g for 10 min at 4°C, and then the supernatant was removed. Cell pellets were sonicated in 200 μL of methanol and centrifuged at 10,000×g for 15 min at 4°C. The methanol extract was stored at −80°C until HPLC analysis was performed. Intracellular uptake of FITC-PLGA NP by HCEC was examined after 4 h by a Leica DMI3000 fluorescence microscope using a 494-nm excitation wavelength.
Cytotoxicity Studies
HCEC were incubated in 96-well transparent plates at 10,000 cells per well (100 μL). After 24 h, the old medium was removed and cells were incubated for 24 and 48 h in the media which contained HB-loaded NP suspension at an equivalent HB NP concentration of 100, 200, and 500 μg/mL. NP were sterilized with UV irradiation 1 day prior to use. Blank culture medium was used as the control. At specified time intervals, the medium was aspirated and rinsed with DPBS twice. Twenty microliters of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) stock solution was then added to each well. Cells were incubated for 4 h at 37°C. Cell viability was measured by absorbance at 485 nm (reference at 590 nm) on an automated plate reader (Bio-Rad, Hercules, CA). The absorbance is directly proportional to the number of living cells in the culture, and the relative cell viability (%) compared to control cells was calculated according to Eq. 8:
Abssample denotes the absorbance of cells incubated with different samples, Abscontrol represents the absorbance of cells incubated with the culture medium only, and Absblank is the absorbance of culture medium without cells.
Statistical Analysis of Data
All studies were performed at least in triplicate. Data were expressed as the mean with 95% confidence intervals. Statistical tests were performed with Student’s t test. For all tests, P values less than 0.05 were considered to be statistically significant. All statistical tests were two tailed.
RESULTS AND DISCUSSION
Drug Encapsulation Efficiency (EE) and Drug Loading (DL)
EE of NP is important for clinical applications as more NP would be required for a given dose if EE is poor. As previously published from our laboratory, the optimized HB-loaded NP formulation under surfactant PVA demonstrated 90.60 ± 2.14% EE and 64.35 ± 1.52% DL [33]. Table I shows EE and DL of NP formulation with different surfactants used in the NP formulation process. The results suggest 76.30 ± 1.84% and 82.52 ± 2.11% of encapsulation efficiency and 54.19 ± 1.31% and 58.60 ± 1.50% of drug loading, respectively, for pluronic- and chitosan-emulsified NP. It is clear that PVA-emulsified NP after optimization possess the highest entrapment efficiency and drug loading among the three formulations, which is probably attributable to high molecular weight of PVA and the optimization process in a previous study.
The apparent final yield (%), which could be calculated as a percentage of the total amount of freeze-dried NP sediments in the total amount of initial materials, was 89.36 ± 2.85%, 91.27 ± 3.28%, and 93.24 ± 4.26% (n = 3), respectively, for the three different NP formulations.
Particle Size and Size Distribution
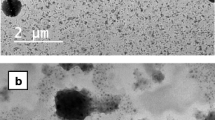
The optimized PVA-, pluronic-, and chitosan-emulsified PLGA NP exhibited 164.3 ± 7.4, 113.4 ± 5.7, and 187.3 ± 9.2 nm of particle size, respectively. Both the optimized NP and unloaded NP demonstrated similar particle sizes. Corresponding SEM studies showed discrete NP with the spherical shape (Fig. 1). NP in the range of 100–200 nm with low polydispersity may be desirable due to high cellular uptake ([34]: 15585275). Surfactant can facilitate NP formulation by lowing surface tension, since less surface energy may be needed to form NP at a given surface area. Small particles of diameter less than 100 nm may not be certainly in favor of ideal NP drug formulation. First, NP of too small particle size have high surface area, which may lead to low EE% with high drug loss into the aqueous phase during preparation ([35]: 11943393; [36]: 14559017). Second, cellular internalization of NP is a process where the curvature of a piece of the lipid bilayer membrane continues to increase and finally form a shell on the NP surface in the endocytosis process. During this process, surface energy of NP contributes to provide the bending energy. It has been reported that NP of diameter <100 nm cannot provide sufficient surface energy to support the required bending energy to form the curvature of the forming size ([34]: 15585275). Computer simulation of NP internalization process further confirmed this mechanism ([37]: 17363051).

Particle size distribution curves and scanning electron photographs of NP. a, b PVA-emulsified HB-loaded PLGA NP; c, d PF-emulsified HB-loaded PLGA NP; and e, f chitosan-emulsified HB-loaded PLGA NP
Surface Charge
Surface charge is an important indication of the stability of a colloidal system in medium as well as for their interaction with cells in vivo. It is known that topical administration of PLGA NP with a charge from ±10 to ±30 mV indicates incipient instability, whereas ±30 to ±40 mV represents moderate stability. The zeta potential and size of unloaded and HB-loaded NP did not differ significantly. Both the optimized and unloaded NP demonstrated similar negative zeta potentials (−20.0 and −20.4 mV, n = 3, respectively). The zeta potential values of PLGA NP with PF-108 and chitosan were schematically presented as −26.9 and +31.0 mV, respectively, which demonstrated larger absolute value of the negative and positive charge. The charge of NP could be due to both the polymers and surfactant. Although pluronic is neutral, anions can bind to the neutral polymers. Pluronic-coated NP may still exhibit nonzero mobility in an external electric field leading to higher negative charge. The polycationic nature of chitosan can help to neutralize the negative charge of polymeric particles. As a result, it leads to strong interactions with lipids on the biological membrane displaying the opposite charge. The values prove the good stability of NP suspended in aqueous medium.
DSC and XRD
To investigate the physical state of HB-loaded in PLGA NP, DSC analysis was performed on polymer, blank, HB, HB-loaded NP, and the physical mixture of HB and blank NP. Drug substance may exist either as a solid solution or solid dispersion (as an amorphous state) or crystalline state in the polymeric matrix. The pure HB showed a single sharp endothermic melting peak at 208.69°C (Fig. 2(E)), which is shifted to a lower temperature 166.32°C (Fig. 2(D)) for HB-loaded NP. However, the HB melting peak totally disappeared in the curves of HB-loaded NP, indicating the absence of crystalline drug in the samples. The polymer may have inhibited the crystallization of HB during NP formulation. Hence, it can be concluded that HB formulated in the NP was in an amorphous or disorderly crystalline phase of a molecular dispersion or a solid solution state in polymeric NP.

DSC thermograms of A polymer, B blank PLGA NP, C HB NP, D mixture, and E drug
An increase in the polymer glass transition temperature (T g) values (49.61°C, Fig. 2(B)) of blank NP was observed relative to the PLGA polymers (45.04°C, Fig. 2(A)). It may be due to rapid solidification of the polymer during the evaporation step of the preparation process. An increase in T g values of HB-loaded NP (53.59°C, Fig. 2(C)) and physical mixture of HB and blank NP (51.22°C, Fig. 2(D)) was observed relative to blank NP. This result indicates the effect of HB on polymers. In addition, XRD analysis was applied to determine the crystalline structure of HB in the NP. The graphs depicted in Fig. 3 show the XRD patterns of blank, HB-loaded NP, and the physical mixture of HB and blank NP. It appeared that blank NP were in an amorphous state (Fig. 3(A)). The crystal diffraction peak of HB was still visible in the mixture of HB and blank NP (Fig. 3(C)). Nevertheless, no crystalline diffraction pattern was distinguishable in the graph of HB-loaded NP (Fig. 3(B)), indicating that HB was present in the NP in an amorphous state, which was consistent with the results of DSC analysis.

X-ray diffractograms of A blank NP, B HB-loaded NP, and C the physical mixture of HB and blank NP
In Vitro Drug Release Profile
In vitro drug release profile of the HB-loaded PLGA NP is shown in Fig. 4. During the initial stage, drug HB loosely bound to or embedded on particle surfaces are released upon contact with bulk release media. Then hydrophobic HB in the external layer of NP may partition through the polymer phase to the bulk aqueous phase. Next, drug in the core of the NP matrix may diffuse out through the hydrophobic polymer matrix. Also during this stage, the polymer may become gradually swollen due to water absorption, further facilitating drug diffusion through the polymer matrix. The small size of the NP is attributable to the release of the majority of drug before the polymer matrix erodes and bulk particle degradation occurs [38]. An initial burst in all release profiles was observed with about 66.1, 60.7, and 70.8% release in the first 24 h for PVA-, pluronic-, and chitosan-emulsified NP, respectively, indicating that HB was loaded most efficiently in chitosan-emulsified NP, followed by PVA- and pluronic- emulsified NP. In a perfect world, drug release from NP formulation would be zero order with continuous drug release over a period of time depending on the drug and indication of interest. However, surface area-to-volume ratio of NP in small size is very large and can lead to burst release of the majority of encapsulated drug over the first few hours and little release afterwards. It was reported that the burst release diminished with increasing particle size and a prolonged release could be obtained when the particles were sufficiently large [39]. The initial burst may be helpful to treat inflammation in a short time. In the following 10 days, the cumulative release percent reached 90.7, 89.6, and 92%, respectively. The release occurs in a sustained manner (sustained release phase) with diffusion from the matrix or polymer degradation, which provides the possibility to continually provide anti-inflammatory response. The cumulative release percent almost achieved 100% after 14–24 days, showing a complete release ability of NP formulation. The generally sustained and controlled release profile of HB facilitates the application of NP for the delivery of corticosteroids. Rather, the release rate of HB from pluronic-emulsified PLGA NP is slowest relative to chitosan-emulsified NP.

Percent cumulative release profiles of HB from a PVA-, PF-, and chitosan-emulsified HB-loaded NP and b thermosensitive gels and PVA-, PF-, and chitosan-emulsified HB-loaded NP suspended in gels. Each data point is expressed as mean ± SD (n = 3)
In vitro release data from NP was fitted to first-order and Higuchi model to describe the pattern of drug release. The first-order release kinetics is directly proportional to the drug concentration involved in the process. The Higuchi model describes the release of HB from an insoluble matrix as the square root of a time-dependent process based on Fickian diffusion. The release rate parameters along with regression values (F, P, and R 2) are displayed in Table II. The regression coefficients with P value <0.05 are highly significant. An optimum model was selected based on the correlation value of various models. The Higuchi model exhibited higher correlation coefficient and linearity than the first-order model. The Higuchi equation indicated the release rate constants of drug from PVA-, pluronic-, and chitosan-emulsified PLGA NP as 4.130, 3.616, and 4.359 h−1/2, respectively. The Korsmeyer-Peppas model describes the release from a polymeric system. To determine the mechanism of drug release, first, 60% drug release data was fitted in the Korsmeyer-Peppas equation and high linearity was observed. The diffusion exponent (n value) was found to be 0.327, 0.365, and 0.309, respectively, for PVA-, pluronic-, and chitosan-emulsified NP. The exponent indicated that the release mechanism from PLGA NP was Fickian in nature, as the n value was less than 0.43 for spherical shape [40, 41]. There are several simultaneous processes including diffusion of water into the matrix, swelling of the matrix due to water entering, formation of gel, diffusion of drug, and dissolution of the polymer matrix. It can be concluded that the release patterns of HB from PVA-, pluronic-, chitosan-emulsified PLGA NP followed the Higuchi model and Fickian diffusion mechanism.
Profiles of drug release from the formulations prepared by dispersing PVA-, pluronic-, and chitosan-emulsified PLGA NP in thermosensitive gel also followed similar kinetics. Triblock copolymer PLGA-PEG-PLGA was synthesized and characterized in our laboratory and a 23% w/w solution was employed as a vehicle for suspending PLGA NP. Ocular administration of such nanoparticulate formulations can result in spontaneous gelation at ocular surface temperature ranging from 34 to 37°C. HB release studies from the gel revealed that the gel structure may not be effective for sustained drug release in vitro. When HB (200 μg) alone was dispersed in the thermosensitive gel, 70% of drug is released in 1 week suggesting negligible resistance of the gel structure on prodrug diffusion (Fig. 4). However, NP dispersed in gels produce none or minimal burst release of drug. An elimination of such initial burst and slower long-term drug release from PLGA NP suspended in gels may be due to diffusional resistance of the gel in a semisolid matrix and/or polymeric adhesion of the gel structure. The thickness of the diffusion boundary layer arises before the parent molecules are released into the aqueous buffer. Thermosensitive gels consist of three-dimensional polymeric networks with excellent water-absorbing capacity and biocompatibility. The gels with great swelling capacity can entrap hydrophobic drugs and thus release them in a controlled fashion by diffusion through and/or by erosion of the gel layer [42]. The studies about the swelling behavior of gels have reported an initial rapid hydration followed by a leveling off. A greater degree of swelling can be achieved in the high molecular weight gels because high molecular weight gels have a higher number of PEG chains and, therefore, a greater ability to expand their network structure. Drug release data of drug HB suspended in gels were used for fitting it to the Fickian and non-Fickian diffusion, resulting in values for n greater than 0.5, suggesting non-Fickian diffusion. Non-Fickian diffusion is desirable, as it indicates that the media penetration rate is in the same range as drug diffusion [43].
The profile indicates a linear zero-order release behavior over 28, 32, and 36 days, respectively, for PVA-, pluronic-, and chitosan-emulsified PLGA NP in thermosensitive gel. The release rates are independent of drug concentration. Release rate constants from NP suspended in gels are observed to be 0.132, 0.120, and 0.144 μg/h for PVA-, pluronic-, and chitosan-emulsified NP, respectively (Table II). The regression coefficients with P value <0.05 are highly significant. The zero-order model exhibited very high regression values (R 2 > 0.95). An association of PLGA blocks in the triblock gel with PLGA NP may exert a behavior effect to prevent burst release. Therefore, dispersion of NP inside the thermosensitive gels may be an effective alternative as controlled release ocular drug delivery systems.
The Korsmeyer-Peppas equation was also applied to determine the mechanism of drug release from the formulations of NP suspended in gels. The n values were calculated to be 0.561, 0.552, and 0.530, which indicated the non-Fickian (anomalous) release for all three formulations, i.e., 0.43 < n < 0.89 [44]. Non-Fickian release refers to the combination of both diffusion and erosion mechanisms of controlled release. The Korsmeyer-Peppas model also showed high correlation for all formulations (Table II). From these results, we concluded that drug release from NP suspended in gels appears to be governed by two phenomena: erosion-based degradation and drug diffusion (non-Fickian diffusion). NP suspended in PLGA-PEG-PLGA thermosensitive gels may result in longer residence time at the site of administration.
In Vitro Cellular Uptake
Cellular uptake of HB-loaded NP was examined qualitatively to visualize the internalization of PVA-, pluronic-, and chitosan-emulsified PLGA NP by laser scanning microscope images of the HCEC. Cells were washed with fresh medium to remove the membrane-bound NP. Intracellular accumulation of FITC-labeled NP was observed and the fluorescence intensity observed in cells increased notably after incubation for 4 h (Fig. 5). Fluorescence images and their superimposition on images demonstrate the interaction of the NP with cells and their intracellular accumulation around the nuclei and most probably into intracellular vacuoles. In addition, the fluorescent chitosan-emulsified NP appear to be internalized by cells more than fluorescent PVA- and pluronic-emulsified NP. No morphological alternation of the cells was detected under the microscope.

Fluorescence microscope images of HCEC cells treated with FITC-labeled a PLGA/PVA NP, b PLGA/PF NP, and c PLGA/CS NP at 3 h
A quantitative investigation has also been conducted by measuring the amount of drug taken up by HCEC. Free drug molecule within the cells was collected in the supernatant by centrifugation. The drug was measured after dissolving NP and sedimenting cell pellets and polymer. The amount of drug was confirmed by the HPLC method. Cellular uptake of NPs by various cell lines appears to be time and dose dependent. Fluorescent PVA-, pluronic-, and chitosan-emulsified PLGA NP (100 μg/mL) were detected in HCEC after 1, 2, 3, and 4 h of incubation at 37°C, respectively. It was observed that the optimum time required for particle uptake at 100 μg/mL was 4 h (Fig. 6a). It is apparent that the percentage of uptake in HCEC after 4 h was 59.09 ± 6.21% for PVA-emulsified NP in comparison with 55.74 ± 6.26% for pluronic-emulsified NP and 62.54 ± 3.30% for chitosan-emulsified NP. It was also found that cellular uptake saturated at a NP concentration of 500 μg/mL (Fig. 6b). Cellular uptake and intracellular accumulation of the substrate notably increased during the first 3 h, followed by a slower. About 71.33, 73.68, and 77.58% of drug were taken up by HCEC within 4 h at a NP concentration of 500 μg/mL for PVA-, pluronic-, and chitosan-emulsified NP, respectively (Fig. 6b). There are multiple pathways of NP accumulation inside cells. For example, under in vivo condition, drug released from NP can penetrate through the cell membrane due to its hydrophobicity [45]. Also, NP may be directly taken up by cells and then retained [46].

a Time-dependent uptake (500 μg/mL) and b dose-dependent uptake of PVA-emulsified PLGA NP (brown bars), PF-emulsified PLGA NP (pink bars), and CS-emulsified PLGA NP (blue-green bars). Each data point is expressed as mean ± SD (n = 3)
The phenomenon of particle size-dependent uptake has been a subject of study. However, the results of their studies differ [47–49]. Most researchers agreed that the shortest wrapping time depends on the particle size. However, the receptor-mediated endocytosis of viruses suggests an optimal size to be 50 nm [50]. Other studies reported maximum uptake of NP with 100 nm particles [34]. These results indicate that interactions between NP and cells probably vary dramatically, resulting in different optimal sizes for cellular uptake.
In Vitro Cell Cytotoxicity
HCEC viability was investigated as a function of NP concentration at 100, 200, and 500 μg/mL after 24 and 48 h using the MTS assay for PVA-, pluronic-, and chitosan-emulsified NP. Cell viability after 24 h did not show significant cytotoxicity for PVA- and pluronic-emulsified NP within the concentration of 100 to 500 μg/mL, while chitosan-emulsified NP demonstrated significant cytotoxicity around the concentration of 500 μg/mL (Fig. 7). After 48 h, NP with surfactants PVA and pluronic still show no significant cytotoxicity, while NP with chitosan indicated significant cytotoxicity within the concentration of 200 and 500 μg/mL with viability of 94.58 ± 1.47% and 84.03 ± 2.46%, respectively. Huang et al. [51] reported that there was no difference in cytotoxicity between chitosan and chitosan nanoparticles. Both chitosan entities exhibited significant cytotoxicity only at concentrations higher than 0.741 mg/mL.

Representation of viability of NP incubated with various concentrations. a HCEC at 24 h and b HCEC at 48 h. Each data point is expressed as mean ± SD (n = 3)
CONCLUSIONS
This study investigates the effect of surfactants such as PVA, pluronic, and chitosan on the entrapment efficiency, particle size, entrapment, and drug loading for HB-loaded NP. The application of thermosensitive gel may eliminate the burst release phase and extend the release rate of HB from NP. Chitosan-emulsified NP showed the highest cellular uptake efficiency relative to PVA- and pluronic-emulsified NP. However, NP with chitosan indicated significant cytotoxicity at the concentrations of 200 and 500 μg/mL with viability of 94.58 ± 1.47% and 84.03 ± 2.46% after 48 h. NP with surfactants PVA and pluronic showed no significant cytotoxicity. These formulations may form a depot in the cul-de-sac to provide a robust concentration gradient for controlled release and efficient uptake of HB within the cornea following topical administration.
References
Urtti A. Challenges and obstacles of ocular pharmacokinetics and drug delivery. Adv Drug Deliv Rev. 2006;58(11):1131–5.
Gulsen D, Chauhan A. Ophthalmic drug delivery through contact lenses. Invest Ophthalmol Vis Sci. 2004;45(7):2342–7.
Shimo T, Yamazaki M, Noguchi Y, Mukawa A, Kato H, Ito Y. Irritation test of dexamethasone valerate (DV-17) and other steroid ointments in rabbits. Skin and eye primary irritation tests, and skin cumulative irritation test. J Toxicol Sci. 1982;7 Suppl 1:1–13.
Komatsu A, Ohashi K, Oba H, Kakehashi T, Mizushima Y, Shirasawa E, et al. Application of lipid microsphere drug delivery system to steroidal ophthalmic preparation. Jpn J Ophthalmol. 1988;32(1):41–3.
Matheson R, Kempers S, Breneman D, Draelos Z, Johnson CE, Loss R, et al. Hydrocortisone butyrate 0.1% lotion in the treatment of atopic dermatitis in pediatric subjects. J Drugs Dermatol. 2008;7(3):266–71.
Eichenfield L, Ellis CN, Fivenson D, Hebert AA, Dromgoole S, Piacquadio D. Evaluation of adrenal suppression of a lipid enhanced, topical emollient cream formulation of hydrocortisone butyrate 0.1% in treating children with atopic dermatitis. Pediatr Dermatol. 2007;24(1):81–4.
Rajka G, Verjans HL. Hydrocortisone 17-butyrate (Locoid) 0.1% fatty cream versus desonide (Apolar) 0.1% ointment in the treatment of patients suffering from atopic dermatitis. J Int Med Res. 1986;14(2):85–90.
Yip YW, Li Wan Po A, Irwin WJ. Kinetics of decomposition and formulation of hydrocortisone butyrate in semiaqueous and gel systems. J Pharm Sci. 1983;72(7):776–81.
Xie S, Tao Y, Pan Y, Qu W, Cheng G, Huang L, et al. Biodegradable nanoparticles for intracellular delivery of antimicrobial agents. J Control Release. 2014;187:101–17.
Rancan F, Blume-Peytavi U, Vogt A. Utilization of biodegradable polymeric materials as delivery agents in dermatology. Clin Cosmet Investig Dermatol. 2014;7:23–34.
Almeida H, Amaral MH, Lobão P, Silva AC, Loboa JM. Applications of polymeric and lipid nanoparticles in ophthalmic pharmaceutical formulations: present and future considerations. J Pharm Pharm Sci. 2014;17(3):278–93.
Achouri D, Alhanout K, Piccerelle P, Andrieu V. Recent advances in ocular drug delivery. Drug Dev Ind Pharm. 2013;39(11):1599–617.
Sadat Tabatabaei Mirakabad F, Nejati-Koshki K, Akbarzadeh A, Yamchi MR, Milani M, Zarghami N, et al. PLGA-based nanoparticles as cancer drug delivery systems. Asian Pac J Cancer Prev. 2014;15(2):517–35.
Pan Q, Xu Q, Boylan NJ, Lamb NW, Emmert DG, Yang JC, et al. Corticosteroid-loaded biodegradable nanoparticles for prevention of corneal allograft rejection in rats. J Control Release. 2015;201:32–40.
Beddoes CM, Case CP, Briscoe WH. Understanding nanoparticle cellular entry: a physicochemical perspective. Adv Colloid Interface Sci. 2015;218C:48–68.
Shen J, Sun M, Ping Q, Ying Z, Liu W. Incorporation of liquid lipid in lipid nanoparticles for ocular drug delivery enhancement. Nanotechnology. 2010;21(2):025101.
Aksungur P, Demirbilek M, Denkbaş EB, Vandervoort J, Ludwig A, Unlü N. Development and characterization of cyclosporine A loaded nanoparticles for ocular drug delivery: cellular toxicity, uptake, and kinetic studies. J Control Release. 2011;151(3):286–94.
Li J, Guo X, Liu Z, Okeke CI, Li N, Zhao H, et al. Preparation and evaluation of charged solid lipid nanoparticles of tetrandrine for ocular drug delivery system: pharmacokinetics, cytotoxicity and cellular uptake studies. Drug Dev Ind Pharm. 2014;40(7):980–7.
Kovacevic A, Savic S, Vuleta G, Müller RH, Keck CM. Polyhydroxy surfactants for the formulation of lipid nanoparticles (SLN and NLC): effects on size, physical stability and particle matrix structure. Int J Pharm. 2011;406(1–2):163–72.
Moradi M, Yamini Y. Surfactant roles in modern sample preparation techniques: a review. J Sep Sci. 2012;35(18):2319–40.
Langevin D. Complexation of oppositely charged polyelectrolytes and surfactants in aqueous solutions. A review. Adv Colloid Interface Sci. 2009;147–148:170–7.
Alakhova DY, Kabanov AV. Pluronics and MDR reversal: an update. Mol Pharm. 2014;11(8):2566–78.
Kabanov AV, Lemieux P, Vinogradov S, Alakhov V. Pluronic block copolymers: novel functional molecules for gene therapy. Adv Drug Deliv Rev. 2002;54(2):223–33.
Kabanov AV, Batrakova EV, Alakhov VY. Pluronic block copolymers as novel polymer therapeutics for drug and gene delivery. J Control Release. 2002;82(2–3):189–212.
Oh SH, Kim JH, Song KS, Jeon BH, Yoon JH, Seo TB, et al. Peripheral nerve regeneration within an asymmetrically porous PLGA/Pluronic F127 nerve guide conduit. Biomaterials. 2008;29(11):1601–9.
Fenske DB, Palmer LR, Chen T, Wong KF, Cullis PR. Cationic poly(ethyleneglycol) lipids incorporated into pre-formed vesicles enhance binding and uptake to BHK cells. Biochim Biophys Acta. 2001;1512(2):259–72.
Ryhänen SJ, Säily MJ, Paukku T, Borocci S, Mancini G, Holopainen JM, et al. Surface charge density determines the efficiency of cationic gemini surfactant based lipofection. Biophys J. 2003;84(1):578–87.
Lin BC, Lin PC, Lai YY, Huang SJ, Yeh FC. The maternal and fetal effects of the addition of sufentanil to 0.5% spinal bupivacaine for cesarean delivery. Acta Anaesthesiol Sin. 1998;36(3):143–8.
Calvo P, Remuñan-López C, Vila-Jato JL, Alonso MJ. Chitosan and chitosan/ethylene oxide-propylene oxide block copolymer nanoparticles as novel carriers for proteins and vaccines. Pharm Res. 1997;14(10):1431–6.
De Campos AM, Sánchez A, Gref R, Calvo P, Alonso MJ. The effect of a PEG versus a chitosan coating on the interaction of drug colloidal carriers with the ocular mucosa. Eur J Pharm Sci. 2003;20(1):73–81.
Andrés E, Albesa-Jové D, Biarnés X, Moerschbacher BM, Guerin ME, Planas A. Structural basis of chitin oligosaccharide deacetylation. Angew Chem Int Ed Engl. 2014;53(27):6882–7.
Wittaya-areekul S, Kruenate J, Prahsarn C. Preparation and in vitro evaluation of mucoadhesive properties of alginate/chitosan microparticles containing prednisolone. Int J Pharm. 2006;312(1–2):113–8.
Yang X, Patel S, Sheng Y, Pal D, Mitra AK. Statistical design for formulation optimization of hydrocortisone butyrate-loaded PLGA nanoparticles. AAPS PharmSciTech. 2014;15(3):569–87.
Win KY, Feng SS. Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials. 2005;26(15):2713–22.
Mu L, Feng SS. Vitamin E TPGS used as emulsifier in the solvent evaporation/extraction technique for fabrication of polymeric nanospheres for controlled release of paclitaxel (Taxol). J Control Release. 2002;80(1–3):129–44.
Ruan G, Feng SS. Preparation and characterization of poly(lactic acid)-poly(ethylene glycol)-poly(lactic acid) (PLA-PEG-PLA) microspheres for controlled release of paclitaxel. Biomaterials. 2003;24(27):5037–44.
Decuzzi P, Ferrari M. The role of specific and non-specific interactions in receptor-mediated endocytosis of nanoparticles. Biomaterials. 2007;28(18):2915–22.
Soppimath KS, Aminabhavi TM, Kulkarni AR, Rudzinski WE. Biodegradable polymeric nanoparticles as drug delivery devices. J Control Release. 2001;70(1–2):1–20.
Zur Mühlen A, Schwarz C, Mehnert W. Solid lipid nanoparticles (SLN) for controlled drug delivery—drug release and release mechanism. Eur J Pharm Biopharm. 1998;45(2):149–55.
Peppas NA. Analysis of Fickian and non-Fickian drug release from polymers. Pharm Acta Helv. 1985;60:110–1.
Lopez-Gasco P, Iglesias I, Benedi J, Lozano R, Teijón JM, Blanco MD. Paclitaxel-loaded polyester nanoparticles prepared by spray-drying technology: in vitro bioactivity evaluation. J Microencapsul. 2011;28:417–29.
Diramio JA, Kisaalita WS, Majetich GF, Shimkus JM. Poly(ethylene glycol) methacrylate/dimethacrylate hydrogels for controlled release of hydrophobic drugs. Biotechnol Prog. 2005;21(4):1281–8.
Kortesuo P, Ahola M, Kangas M, Yli-Urpo A, Kiesvaara J, Marvola M. In vitro release of dexmedetomidine from silica xerogel monoliths: effect of sol-gel synthesis parameters. Int J Pharm. 2001;221(1–2):107–14.
Duvvuri S, Janoria KG, Mitra AK. Development of a novel formulation containing poly(D,L-lactide-coglycolide) microspheres dispersed in PLGA-PEG-PLGA gel for sustained delivery of ganciclovir. J Control Release. 2005;108(2–3):282–93.
Zhang L, Pornpattananangku D, Hu CM, Huang CM. Development of nanoparticles for antimicrobial drug delivery. Curr Med Chem. 2010;17(6):585–94.
Akinc A, Battaglia G. Exploiting endocytosis for nanomedicines. Cold Spring Harb Perspect Biol. 2013;5(11):a016980.
Choi JS, Cao J, Naeem M, Noh J, Hasan N, Choi HK, et al. Size-controlled biodegradable nanoparticles: preparation and size-dependent cellular uptake and tumor cell growth inhibition. Colloids Surf B: Biointerfaces. 2014;122:545–51.
Zhu J, Liao L, Zhu L, Zhang P, Guo K, Kong J, et al. Size-dependent cellular uptake efficiency, mechanism, and cytotoxicity of silica nanoparticles toward HeLa cells. Talanta. 2013;107:408–15.
Yue T, Zhang X. Cooperative effect in receptor-mediated endocytosis of multiple nanoparticles. ACS Nano. 2012;6(4):3196–205.
Lu F, Wu SH, Hung Y, Mou CY. Size effect on cell uptake in well-suspended, uniform mesoporous silica nanoparticles. Small. 2009;5(12):1408–13.
Huang M, Khor E, Lim LY. Uptake and cytotoxicity of chitosan molecules and nanoparticles: effects of molecular weight and degree of deacetylation. Pharm Res. 2004;21(2):344–53.
Acknowledgments
We sincerely thank Dr. James B Murowchick from the Department of Geosciences at the University of Missouri-Kansas City for his assistance with X-ray pictures of compounds and NP. This work was supported by Missouri Life Sciences Research Grant No. MLSRB0017025.
Conflict of Interest
No competing financial interests exist.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yang, X., Trinh, H.M., Agrahari, V. et al. Nanoparticle-Based Topical Ophthalmic Gel Formulation for Sustained Release of Hydrocortisone Butyrate. AAPS PharmSciTech 17, 294–306 (2016). https://doi.org/10.1208/s12249-015-0354-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12249-015-0354-5