
Abstract
Accurate prediction of drug-drug interactions (DDI) is a challenging task in drug discovery and development. It requires determination of enzyme inhibition in vitro which is highly system-dependent for many compounds. The aim of this study was to investigate whether the determination of intracellular unbound concentrations in primary human hepatocytes can be used to bridge discrepancies between results obtained using human liver microsomes and hepatocytes. Specifically, we investigated if Kpuu could reconcile differences in CYP enzyme inhibition values (Ki or IC50). Firstly, our methodology for determination of Kpuu was optimized for human hepatocytes, using four well-studied reference compounds. Secondly, the methodology was applied to a series of structurally related CYP2C9 inhibitors from a Roche discovery project. Lastly, the Kpuu values of three commonly used CYP3A4 inhibitors—ketoconazole, itraconazole, and posaconazole—were determined and compared to compound-specific hepatic enrichment factors obtained from physiologically based modeling of clinical DDI studies with these three compounds. Kpuu obtained in suspended human hepatocytes gave good predictions of system-dependent differences in vitro. The Kpuu was also in fair agreement with the compound-specific hepatic enrichment factors in DDI models and can therefore be used to improve estimations of enrichment factors in physiologically based pharmacokinetic modeling.
Similar content being viewed by others

Introduction
Accurate predictions of drug-drug interactions (DDIs) are a challenging task during drug development because the relevant inhibitor concentration is not directly accessible (1,2). Predictions of DDIs depend on in vitro parameters such as the half-maximal inhibitory concentration (IC50) or the inhibition constant (Ki) (3). These in vitro values are then used in physiologically based pharmacokinetic (PBPK) models to simulate DDIs in vivo (4).
For predictions of metabolic DDIs with CYP enzymes, Ki or IC50 values for a given compound are typically determined in liver microsomes or in hepatocytes. There is a growing interest in using cryopreserved human hepatocytes (HH) instead of human liver microsomes (HLM) for preclinical DDI assessment. In the microsomal setting, the drug concentration available to interact with the enzyme is equal to the unbound drug concentration in the incubation medium (i.e., the nominal incubation concentration, Cinc, corrected for fraction of unbound drug in the liver microsomes (fu,mic)) (5). However, the HH model may be a more physiologically relevant system than HLM, because it takes into account multiple processes that influence enzyme inhibition. In the hepatocyte setting, compounds need to reach the cell interior where the CYP enzymes are located, a process that may involve passive permeability and active transport. Subsequent to this, the intracellular unbound drug concentration (Cu,cell) may be further influenced by metabolic clearance, protein binding, and partitioning into organelles (e.g., lysosomal trapping) or cellular membranes (5,6,7,8). These factors can all contribute to inconsistencies between the Cu,cell and Cinc, leading to differences in the Ki,app or IC50,app values measured in HLM and HH. Therefore, careful consideration must be given to which in vitro system is appropriate for testing enzyme inhibition.
Determination of Cu,cell is especially challenging if active transport is involved and is therefore classified as a low-confidence parameter in PBPK modeling (9). The US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) recommended the [I]/Ki ratio (where [I] is the inhibitor concentration) as a parameter in static DDI predictions (10). However, the determination of [I] is not currently standardized. The surrogates commonly used are the average or maximum unbound concentration in blood or at the inlet to the liver, or the maximum total concentration in the circulation at steady state (10,11,12). In addition to the different methods to estimate [I], the determination of Ki also varies. For example, Ki,app—based on determinations in cryopreserved HH suspended in human plasma—has been proposed (13). Such systems aim to reduce in vitro to in vivo translational uncertainties by more closely mimicking the in vivo situation, building protein binding and intracellular bioavailability considerations into the in vitro system.
Strategies for more direct estimations of Cu,cell recently emerged (14,15,16,17,18). These include the parallel determination of hepatocellular drug accumulation at steady state (Kp) and the unbound fraction of drug in the cell (fu,cell) which are combined to calculate the unbound partition coefficient Kpuu. Kpuu is defined as the ratio between unbound drug concentration in the cell interior and the unbound drug concentration in the cell exterior at steady state and can also be derived from kinetic parameters (17).
We recently introduced the term intracellular bioavailability (Fic), defined as the ratio between the intracellular unbound concentration, which is available to elicit effects inside the cell, and the extracellular concentration. In the present work, Fic is equal to Kpuu, as no protein was added to the incubation media, and therefore, the term Kpuu has been used for simplicity. Fic or Kpuu account for active and passive mechanisms and predict target engagement and phenotypic responses in cells (19). We have shown that this parameter reflects transporter effects in cell lines transfected with single transporters. Further, we have seen differences in the Kpuu values for freshly isolated, plated HH, and suspended HH. This is explained by the altered clearance in the two systems (7).
Given this strong influence of culture formats on Kpuu, we hypothesized that Kpuu could be used to reconcile the differences in measurement obtained from different human-relevant experimental systems. We therefore first investigated a series of reference CYP inhibitors (saquinavir, nelfinavir, enoxacin, and clarithromycin) in HLM and HH, previously shown to have differences in Ki,app in rat liver microsomes and rat hepatocytes (20,21). Second, we applied an optimized assay for a series of investigational compounds from a drug discovery program that were all identified as CYP2C9 inhibitors. Finally, Kpuu was determined for a series of commonly used CYP inhibitors of the azole antifungal family—ketoconazole, itraconazole, and posaconazole. These compounds were chosen because they require hepatic enrichment factors (referred to as “hepatic uptake value” in the SimCYP software), to reconcile in vitro and in vivo Ki values (11,22). We reasoned that these scaling factors represent Kpuu at the site of DDI.
Methods
Chemicals
Compounds were retrieved from the in-house stock at the Roche laboratories at their highest available purity and dissolved at 10 mM in DMSO or, if lower, at their highest solubility. DMSO stocks were kept at − 20°C.
Compound Selection
The literature was screened for a validation set of compounds with reported discrepancies between cellular and microsomal IC50 or Ki values. Enoxacin, clarithromycin, saquinavir, and nelfinavir were identified as suitable candidates (20). The method was then applied to an internal compound set from Roche (hereafter RO compounds) consisting of a structurally related series known to inhibit CYP2C9, and for which information on IC50 in HLM and HH was available. As a third compound set, three members of the azole antifungal family—ketoconazole, itraconazole, and posaconazole—were identified as compounds with reported in vitro Ki values that differ from the Ki values reported in models predicting in vivo data.
Cell Culture
A pooled HH batch (BioreclamationIVT, LiverPool 10-donor HH, product no. X008001, Lot RBR) was used in the CYP inhibition assay and for determination drug uptake in suspended HH. After thawing, the cells were suspended in InVitroGro CP medium at the specified concentration in each assay (product no. Z99029, BioreclamationIVT) and used immediately for experiments.
CYP Inhibition Assay in HLM and HH
For determination of CYP2C9 inhibition, 180 μL of HLM 0.2 mg/mL (BD Gentest, Cat no. 452117, pool from 150 donors of mixed gender) plus substrate (5 μM diclofenac for CYP2C9 and 5 μM midazolam for CYP3A4) was added to a deep 96-well plate together with the test compound or reference inhibitor (concentration range from 0.008 μM to 1 μM). After the addition of 20 μL of 10 mM NADPH, the plate was incubated for 5 min. The reaction was stopped by adding 200 μL of acetonitrile containing an internal standard. After centrifugation of 10 min at 6200×g, the concentration of the substrate in the samples was determined by LC-MS (Sciex API4000, see S6 for LC-MS parameters).
For HH, 50 μL of a suspension of cryopreserved HH (3 × 106 cells/mL) were added to a 96-well round bottom plate and pre-incubated for 20 min in Williams E medium (without FBS). Fifty microliters of a × 3 concentrated test compound-solution and 50 μL of × 3 concentrated substrate (diclofenac for CYP2C9, midazolam for CYP3A4 and tacrine for CYP1A2) were added and the plate was incubated on a shaker at 37°C, 5% CO2, and 900 rpm. Incubations were stopped after 5 min by transferring 100 μL of the suspension to a fresh plate and adding 100 μL of acetonitrile containing the internal standard and then analyzed as above.
Where available, IC50 values were compared to values found in the literature (Table S1).
Intracellular Unbound Drug Accumulation Ratio (Kpuu)
Kpuu was determined as previously described (7,18) using Eq. (1):
where Kp is the steady-state cellular uptake, and fu,cell the fraction of unbound compound in the cell, determined as described below. The fraction unbound of the compound in the medium (fu,medium) was equal to 1 as no serum proteins or other drug binding components were added to the incubation media.
Steady-State Cellular Uptake (Kp)
The Kp in suspended HH was measured similarly to previous reports with some modifications (7,18). Briefly, cells were rinsed twice with pre-warmed containing 10 mM HEPES. One hundred microliters of cell suspension (5 × 106 cells/mL) was added to a conical 96-well plate, and 100 μL of 1 μM drug solutions in Hank’s buffered salt solution (HBSS) buffered with 10 mM HEPES was added (resulting in a 0.5 μM final concentration). All incubations were performed in triplicates on up to three independent occasions. Full-time curves were established at 1, 3, 7.5, 15, 30, and 45 min to determine Kp at equilibrium. For single time point measurements, the incubation time was matched to the CYP inhibition assays. A sample of the medium was removed after centrifugation in a pre-cooled centrifuge at 4°C (100×g, 5 min), and ten-fold diluted with a mixture of acetonitrile and water (60:40) containing the internal standard for determination of Cmedium. The remaining supernatant was removed by aspiration and cells were washed twice with ice-cold buffer. The intracellular compound was extracted using the acetonitrile to water to internal standard mixture for determination of the amount of drug in the cells (Acell). Protein content (Pcell) was quantified using the BCA assay in representative wells to establish the cellular volume (Vcell), assuming 6.5 μL/mg protein (23). Finally, Kp was calculated using Eq. (2)
Determination of fu,cell and fu,mic
fu,cell was measured in cassette mode as previously described, but with minor modifications (24). Briefly, frozen cell pellets were thawed on ice and diluted to 10 million cells/mL in HBSS containing 10 mM HEPES. The suspension was homogenized using a mini bead beater (Precellys, EQ02520-300-RD000.0, bead no. VK01) for 2 cycles of 10 s with an intermittent time of 30 s. Compounds were added to the cell homogenate at a final concentration of 0.5 μM and the spiked homogenate was transferred to a dialysis chamber (Rapid Equilibrium Dialysis Device, Thermo Fisher Scientific). HBSS buffered with 10 mM HEPES was placed in the receptor compartment. Samples of spiked cell homogenate were placed at 4 and 37°C for the duration of the experiment for recovery and stability calculations. Incubation time was 4 h at 37°C and 900 rpm for all compounds except itraconazole which reached equilibrium only after 24 h (Fig. S4). The unbound fraction in the cell homogenate (fu,hom) was determined according to Eq. (3):
and the fraction of unbound compound in the cell (fu,cell) was calculated by correcting for homogenate dilution according to Eq. (4):
where the dilution constant D was calculated using Eq. (5), and assuming the Vcell to be equal to 6.5 μL/mg protein (23). Phom is the protein concentration of the cell homogenate quantified using the BCA assay.
The lower detection limit was reached for determination of fu,cell below 0.01%, and the binding was assumed to be linear at the concentrations used, based on previous results (18).
For determination of the fraction unbound to microsomes (fu,mic), HLM at 0.2 mg/mL (equivalent concentration to CYP inhibition assay; BD Gentest, Cat no. 452117, pool from 150 donors of mixed gender) were used instead of cell homogenate and calculated in analogy to fu,hom (Eq. 3).
Determination of Molecular Properties
Molecular properties of literature compounds were determined using ADMET predictor (Simulations Plus, version 7.2) and cross-checked to values published in the public databases DrugBank (25) and PubChem (http://pubchem.ncbi.nlm.nih.gov). Molecular properties (logD, charge, PSA) of RO compounds were calculated using Roche proprietary in silico tools.
Statistical Analysis
All statistical analyses were performed in Graph-Pad Prism (version 7.04).
Results
Method Optimization
Four reference compounds of different chemical character were studied first. The physicochemical properties of these drugs are summarized in Table I. Enoxacin and clarithromycin are poorly metabolized, hydrophilic drugs while saquinavir and nelfinavir are extensively metabolized, lipophilic drugs (26). All four have been previously shown to have a clear difference in their Ki,app values for liver microsomes and suspended hepatocytes in the rat (Table S1). This made these compounds suitable for investigation in human liver microsomes (HLM) and hepatocytes (HH). The fraction unbound of the four compounds to HLM (fu,mic) was determined using equilibrium dialysis and compared to fu,mic in rat liver microsomes (Fig. 1a, Table S3). Kpuu in suspended HH was determined by combining Kp and fu,cell (Table I). Kpuu in rat hepatocytes was derived from Kp values from the literature (Table S3). Enoxacin and clarithromycin had Kpuu values above 1, indicating that they accumulated in HH. In contrast, saquinavir and nelfinavir had Kpuu values below 1 (Fig. 1b).

a Comparison of fu,mic in human vs. rat liver microsomes. Rat values are derived from Brown et al. (20). b Comparison of Kpuu in human vs. rat hepatocytes. c Log fold difference of apparent and corrected (unbound) IC50 determined in human liver microsomes and human hepatocytes or Ki determined in rat microsomes and rat hepatocytes. The dotted lines represent a 2-fold difference. Error bars represent standard deviations. Apparent and corrected Ki or IC50 values are presented in Tables S1 and S2. Enox, E: enoxacin; Clar, C: clarithromycin; Saq, S: saquinavir; Nel, N: nelfinavir
Next, we used the fu,mic and Kpuu values in human and rat to scale apparent Ki (or IC50) values determined in microsomes and hepatocytes, respectively. Before scaling, these apparent values revealed a significant discrepancy between hepatocytes and microsomes, with the differences ranging up to 22-fold (Fig. 1c). To obtain corrected Ki or IC50, we multiplied apparent Ki or IC50 in microsomes with fu,mic and apparent Ki or IC50 in hepatocytes with Kpuu (= Kp × fu,cell). After applying these corrections, Ki,app or IC50,app values could largely be reconciled (i.e., ~ 2-fold differences). Nelfinavir—the most lipophilic compound in the series—appeared to be an outlier since after correction for fu,mic and Kpuu, the difference in Ki or IC50 increased rather than decreased.
Kpuu as Scaling Factor of IC50 for an RO Discovery Series Inhibiting CYP2C9
We further applied our approach to an internal compound set (RO compounds). The nine compounds in this discovery series were outliers in the internal screening processes in which IC50 values of HH and HLM were compared. The compounds of this series are known to inhibit CYP2C9. These nine compounds are structurally related, with a common core structure containing a sulfonamide and a secondary amide, and aromatic substituents including (iso-)thiazoles, benzothiophenes, pyridines, or furanes (Fig. 2a). They are all acidic at pH 7.4, with logD values indicating hydrophilic properties (Table II).

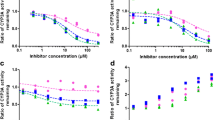
Kpuu as correction factor of IC50 in the RO discovery series. a Structures of RO discovery compound. b–e Comparison of IC50 values measured in HLM and HH. Hepatocellular IC50,app were corrected with Kpuu (= Kp · fu,cell) in suspended HH and microsomal IC50,app values were corrected with fu,mic to obtain IC50,corr. The dotted line indicates the line of unity. The labels of the data points correspond to the structure numbers in panel (a)
First, we determined the IC50,app in HH and HLM using diclofenac as substrate. The IC50 values of RO1, RO3, and RO4 were identical for HH and HLM, while the other compounds differed up to 12-fold (RO9) (Fig. 2b), with, a root-mean-squared error (RMSE) of 9.4 for IC50,hep,app compared to IC50,mic,app. As in the previous section, we next determined Kpuu and fu,mic taking care to use the same batches of hepatocytes and microsomes as for the inhibition experiments, respectively (Table II). All parameters were matched to the IC50 assay.
We then used the Kpuu and fu,mic values as scaling factors to determine IC50,corr. Correction using Kpuu or fu,mic alone did not improve the correlations (Fig. 2c and d). After combining Kpuu and fu,mic to give IC50,hep,corr and IC50mic,corr, respectively, the linear correlation between the IC50 values improved, giving and R2 of 0.92 and a 10-fold reduction in RMSE from 9.4 to 0.9 (Fig. 2e).
Because the compounds were analogous to each other, we also analyzed the possible influence of substituents on Kpuu. For instance, in five of the nine compounds, the R2 substituent was kept constant while the R1 substituent varied. However, no systematic influence of the change in substituent could be observed. We conclude that the compound series was too small to allow a proper substituent analysis; this underscores the importance of experimental analysis of small series of compounds.
Kpuu in IVIVE and DDI Models of the Azole Antifungal Family
Finally, we investigated if Kpuu could improve predictions by DDI models that are based on in vitro Ki values. Compounds of the azole family are commonly used as inhibitors of CYP3A4 in preclinical and clinical DDI studies (2,27). A concentrative cellular uptake of ketoconazole, itraconazole, and posaconazole has been widely described, but uptake mechanisms are not fully elucidated (28,29,30,31). This concentrative uptake requires correction of Ki (which is typically determined in HLM) in order to match in vivo Ki values in DDI models. Therefore, compound-specific correction factors (referred to as “hepatic enrichment factor” or “hepatic uptake scalar”) have been introduced to improve the IVIVE in several models, which are summarized in Fig. 3a. We therefore investigated if Kpuu values measured in suspended HH agreed with these correction factors (Table III and Fig. 3b). The Kpuu values were high, ranging from 7 to 38, in line with the previously described concentrative uptake. Interestingly, the Kpuu values were within 2-fold of the empirically determined correction factors in the DDI models, indicating that Kpuu is a major contributor to the “hepatic enrichment factor” for the azole series.

Correlation of IVIVE correction factors with Kpuu. a Overview of correction factors used for the azole antifungal compounds in in vivo DDI studies. b Illustration of hepatic uptake scalars in rat and human DDI models (error bars represent range) vs. in vitro Kpuu in suspended HH (error bars represent SD)
Discussion
In this work, we evaluated the potential of Kpuu as scaling factor for CYP enzyme inhibition studies, using three experimental setups.
For the first set of compounds, we investigated if the determination of intracellular unbound concentrations could be used to reconcile differences in potency in CYP inhibition observed between microsomes and hepatocytes. CYP enzymes can be considered as targets or off-targets facing the cell interior. We have previously shown that Fic or Kpuu can explain differences in potency between isolated intracellular targets in biochemical high-throughput screening assays and the corresponding cellular assays (19). Drug response for intracellular targets in a variety of subcellular compartments including the cytosol, endosomes, and the nucleus has been successfully predicted, using our methodology for determination of Fic, Fcyto, or Fendo (19,32). Intracellular unbound drug concentrations are determined by combining the cellular accumulation ratio (Kp) with the fraction of unbound compound in the cell (fu,cell). fu,cell is determined using the homogenization method which relies on the assumption that the degree of unspecific binding of drugs is not altered upon homogenization of cells (7,18).
In the first series of experiments, we used reference compounds with previously reported differences in Ki of CYPs in rat liver microsomes and rat hepatocytes, as well as Kp, fu,cell, and fu,mic values in the rat (20).
Our experimental human fu,cell values differed more than 100-fold for the two lipophilic drugs compared to those in the rat study that was calculated based on lipophilicity and cellular volume (33) (saquinavir 0.14 vs. 0.0003 and nelfinavir 0.035 vs. 0.0002, rat vs. human, respectively, Table S3). There was a better agreement for the two more hydrophilic compounds (enoxacin 0.99 vs. 1.00 and clarithromycin 0.86 vs. 0.10 in rat vs. human, respectively). The homogenization/membrane dialysis method has been compared with the temperature and lipophilicity methods for estimating the fu,cell of a limited series of structural unrelated compounds by Riede et al. (15). In their study, the homogenization method gave generally lower values than the temperature and lipophilicity methods. This suggests an overestimation of intracellular binding by the latter two. However, a large variability (up to 13-fold differences) between laboratories has been observed (7,15,17,34,35). fu,cell is dependent on accurate dilution factors for scaling fu,hom to fu,cell (Eq. 4 and 5). In the comparative study by Riede et al. (15), very low cell concentrations (130,000 cells/mL) were used, in comparison to 10 million cells/mL in our studies or 50 million cells/mL by Riccardi et al. (34,35). Such experimental differences may not allow a reliable comparison between laboratories, but in general, all experimental setups agree on significantly lower fu,cell for highly lipophilic drugs as compared to calculations based on logD. This indicates the importance of using a cellular matrix with relevant lipid and protein binding sites of drugs for the determination of fu,cell (8). Moreover, no significant inter-species variability in fu,cell has been observed in a recent comparative study (34). Therefore, for the four reference compounds, the methodological differences were more significant than the expected inter-species variability in fu,cell, and we used our experimental fu,cell values for determination of Kpuu in both rat and human. On the other hand, inter-species variability is expected in Kp values, which can be influenced by active uptake and elimination processes. Therefore, the specific Kp in rat or human hepatocytes was used for determination of Kpuu (Table S3). For the four reference compounds, Kp for the human and rat hepatocytes followed the same rank order and were in the same order of magnitude, with slightly higher values in rat hepatocytes for three out of the four compounds. It should be noted that clarithromycin is a weak base that can be subject to lysosomal trapping. Using pH partitioning theory (18), the calculated cytosolic Kpuu (or Fcyto) in human hepatocytes would result in 1.3, instead of the experimental value of 2.7, which does not account for subcellular localization of the drug.
Two of the four reference compounds, enoxacin and clarithromycin, displayed a Ki ratio (Ki,mic/Ki,hep) or IC50 ratio and Kpuu above one, indicating a concentrative uptake of the compounds in hepatocytes. By contrast, the Ki ratio or IC50 ratio and Kpuu were less than one for saquinavir and nelfinavir, suggesting that the lower potency in rat hepatocytes was driven by poor access to the intracellular compartment (and to the CYP enzyme) as opposed to the microsomes where the CYP enzyme is freely exposed in the medium. For three out of the four compounds, Ki or IC50 values in liver microsomes and hepatocytes were in good agreement after applying relevant Kpuu and fu,mic as scaling factors (Fig. 1c) (34,35,36). For nelfinavir, scaling Ki or IC50 values with Kpuu and fu,mic resulted in a larger discrepancy rather than an improvement. Interestingly, nelfinavir was identified as an outlier also in our previous Kpuu studies in MDCK cells, suggesting the involvement of unknown active processes that are not captured by the Kpuu methodology (7). Nelfinavir is very lipophilic, poorly soluble, and a substrate of active efflux (P-gp)—these properties suggest that the compound is prone to nonspecific binding and that there are significant confounding effects occurring in the hepatocyte experiment. Indeed, poor mass balance (~ 50%) was observed for nelfinavir and the similarly lipophilic saquinavir in the hepatocyte experiments. As the mass balance was recovered in the presence of the CYP inhibitor ABT, this confirms that the loss of compound is due to hepatic metabolism (Fig. S4). The hepatocyte metabolism of nelfinavir and saquinavir has been reported previously in a study that identified these two compounds as outliers. This was based on their intrinsic clearance in microsomes being much higher than in hepatocytes, compared to other compounds with similar clearance mechanisms (37).
It should be noted, however, that in our studies, the hepatocyte metabolism of nelfinavir and saquinavir was not a limiting factor for establishing the concentration equilibrium, since Kp values were unaffected by the presence or absence of ABT (Fig. S4). Furthermore, the use of albumin or an increase in temperature of wash buffers from 4 to 37°C (38,39) did not change the results significantly (Fig. S5), which precludes the likelihood that non-specific binding was affecting the results. Thus, the discrepancy in the Ki,corr values of nelfinavir could not be explained by CYP-mediated metabolism or non-specific binding and is likely driven by other factors. Despite the discrepancy for nelfinavir, we overall obtained a significant harmonization of the Ki ratios between microsomes and hepatocytes, suggesting the validity of this approach and indicating the value of further elaboration.
In the next set of experiments, we therefore harmonized the experimental conditions between the CYP inhibition experiments and the Kpuu experiments. Instead of comparing Kpuu to CYP inhibition constants from the literature, we determined IC50 in HH and HLM with methodologies as similar as possible to each other to generate a consistent dataset. As a result, the Kpuu- and fu,mic-corrected IC50 values for the discovery series of CYP2C9 enzyme inhibitors were in excellent agreement for HH and HLM (Fig. 2e). We conclude that the best agreement between the two systems is obtained by applying simultaneous correction factors for both systems, along with taking into consideration differences in assay setup and inter-batch variability.
Given the significance of Kpuu in the two series of in vitro experiments, we next evaluated if it could also be used as a scaling factor in in vivo studies. More specifically, we investigated if Kpuu could be used to estimate the empirical hepatic uptake scalar required in PBPK models of ketoconazole, itraconazole, and posaconazole as CYP inhibitors. Many models use such empirically determined scalars (Fig. 3a). However, others avoid them because the uptake mechanism of these azole compounds are not mediated via classic hepatic uptake transporters such as the OATP or OCT transporter family (29,40). Kpuu is a measure of all combined effects in a cell and provides a mechanistic tool for estimation of uptake scalars without the need for elucidating the underlying factors contributing to the net drug disposition in the cell. We found that Kpuu values in suspended HH were in good agreement with the empirically determined hepatic uptake scalars in the models for both rat and human (Fig. 3). In the case of itraconazole, the three major metabolites are equally or more potent inhibitors of CYP3A4 than the parent compound, which is a complicating factor in PBPK modeling (28,40,41). The measurement of Kpuu of the parent compound alone was however sufficient for recovering the hepatic uptake scalars. Therefore, we believe that Kpuu can be rationally integrated into prospective PBPK modeling approaches. The use of HH instead of its rat counterpart reduces the potential effects of inter-species differences in drug transporter expression and metabolic enzyme sensitivity to inhibition. It is also the most relevant in vivo model system for making extrapolations to humans. The relevance of Kpuu to in vivo studies is further supported by a recent study in which a series of four statins was assessed for in vitro and in vivo liver enrichment (35); in vivo liver-to-plasma, Kpuu in rat and human were in good agreement with in vitro Kpuu obtained in cryopreserved suspended hepatocytes. Similar scenarios are anticipated for, e.g., rifampicin, which has an empirical hepatic uptake scalar of 16.9 (42) and for bosentan, with a hepatic uptake scalar of 5 to 6 (43).
Determination of Ki,app directly in HH can be an alternative to the incorporation of hepatic uptake scalars. However, most Ki screens are based on HLM. To make our approach more generally applicable, we suggest the use of Ki,corr values, and account for intracellular unbound concentrations in the target cells in any tissue of interest (Fig. 4). Our approach is not limited to the liver and can be applied on any cell of interest with an intracellular target, including studies of DDIs in intestinal enterocytes.

Summary of use and interpretation of Kpuu in the context of CYP-mediated DDI
Conclusion
In summary, our results indicate that Kpuu in HH is an easy-to-interpret in vitro parameter that can be used as a human-relevant scaling factor to bridge the differences of experimental systems such as liver microsomes and hepatocytes and gives a mechanistic understanding of any result discrepancies (Fig. 4). Since Kpuu is the net result of all processes that affect the intracellular unbound drug concentration in HH, no prior knowledge of uptake and elimination processes is required. Indeed, very high or low Kpuu values can indicate that active cellular processes are playing a significant role in drug disposition. Therefore, Kpuu can be used as a decision tool (e.g., if the Kpuu is lower than 0.5 or higher than 2) for more detailed investigations. Very lipophilic compounds have been found to be more challenging for assessment of Kpuu and future work should focus on this compound class. Further, Kpuu has been shown to reflect hepatic uptake scalars used in literature for the triazole antifungal family and we believe that the use of Kpuu is an approach that can be pursued to provide a mechanistic understanding of scalars used in PBPK models that predict drug exposure and DDI potential.
References
Obach RS. Predicting drug-drug interactions from in vitro drug metabolism data: challenges and recent advances. Curr Opin Drug Discov Dev. 2009;12(1):81–9.
Min JS, Bae SK. Prediction of drug-drug interaction potential using physiologically based pharmacokinetic modeling. Arch Pharm Res. 2017;40(12):1356–79.
Sekiguchi N, Higashida A, Kato M, Nabuchi Y, Mitsui T, Takanashi K, et al. Prediction of drug-drug interactions based on time-dependent inhibition from high throughput screening of cytochrome P450 3A4 inhibition. Drug Metab Pharmacokinet. 2009;24(6):500–10.
Zhuang X, Lu C. PBPK modeling and simulation in drug research and development. Acta Pharm Sin B. 2016;6(5):430–40.
Hewitt NJ, Lechon MJ, Houston JB, Hallifax D, Brown HS, Maurel P, et al. Primary hepatocytes: current understanding of the regulation of metabolic enzymes and transporter proteins, and pharmaceutical practice for the use of hepatocytes in metabolism, enzyme induction, transporter, clearance, and hepatotoxicity studies. Drug Metab Rev. 2007;39(1):159–234.
Cantrill C, Houston JB. Understanding the interplay between uptake and efflux transporters within in vitro systems in defining hepatocellular drug concentrations. J Pharm Sci. 2017;106(9):2815–25.
Mateus A, Treyer A, Wegler C, Karlgren M, Matsson P, Artursson P. Intracellular drug bioavailability: a new predictor of system dependent drug disposition. Sci Rep. 2017;7:43047.
Treyer A, Mateus A, Wisniewski JR, Boriss H, Matsson P, Artursson P. Intracellular drug bioavailability: effect of neutral lipids and phospholipids. Mol Pharm. 2018;15(6):2224–33.
Jones HM, Chen Y, Gibson C, Heimbach T, Parrott N, Peters SA, et al. Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin Pharmacol Ther. 2015;97(3):247–62.
Iwasaki S, Hirabayashi H, Funami M, Amano N. Unbound liver concentration is the true inhibitor concentration that determines cytochrome P450-mediated drug-drug interactions in rat liver. Xenobiotica. 2017;47(6):488–97.
Kato M, Shitara Y, Sato H, Yoshisue K, Hirano M, Ikeda T, et al. The quantitative prediction of CYP-mediated drug interaction by physiologically based pharmacokinetic modeling. Pharm Res. 2008;25(8):1891–901.
Lu C, Miwa GT, Prakash SR, Gan LS, Balani SK. A novel model for the prediction of drug-drug interactions in humans based on in vitro cytochrome p450 phenotypic data. Drug Metab Dispos. 2007;35(1):79–85.
Mao J, Mohutsky MA, Harrelson JP, Wrighton SA, Hall SD. Prediction of CYP3A-mediated drug-drug interactions using human hepatocytes suspended in human plasma. Drug Metab Dispos. 2011;39(4):591–602.
Riccardi K, Li Z, Brown JA, Gorgoglione MF, Niosi M, Gosset J, et al. Determination of unbound partition coefficient and in vitro-in vivo extrapolation for SLC13A transporter-mediated uptake. Drug Metab Dispos. 2016;44(10):1633–42.
Riede J, Camenisch G, Huwyler J, Poller B. Current in vitro methods to determine hepatic Kpuu: a comparison of their usefulness and limitations. J Pharm Sci. 2017;106(9):2805–14.
Yoshikado T, Toshimoto K, Nakada T, Ikejiri K, Kusuhara H, Maeda K, et al. Comparison of methods for estimating unbound intracellular-to-medium concentration ratios in rat and human hepatocytes using statins. Drug Metab Dispos. 2017;45(7):779–89.
Guo Y, Chu X, Parrott NJ, Brouwer KLR, Hsu V, Nagar S, et al. Advancing predictions of tissue and intracellular drug concentrations using in vitro, imaging and PBPK modeling approaches. Clin Pharmacol Ther. 2018.
Mateus A, Matsson P, Artursson P. Rapid measurement of intracellular unbound drug concentrations. Mol Pharm. 2013;10(6):2467–78.
Mateus A, Gordon LJ, Wayne GJ, Almqvist H, Axelsson H, Seashore-Ludlow B, et al. Prediction of intracellular exposure bridges the gap between target- and cell-based drug discovery. Proc Natl Acad Sci U S A. 2017;114(30):E6231–9.
Brown HS, Wilby AJ, Alder J, Houston JB. Comparative use of isolated hepatocytes and hepatic microsomes for cytochrome P450 inhibition studies: transporter-enzyme interplay. Drug Metab Dispos. 2010;38(12):2139–46.
Parker AJ, Houston JB. Rate-limiting steps in hepatic drug clearance: comparison of hepatocellular uptake and metabolism with microsomal metabolism of saquinavir, nelfinavir, and ritonavir. Drug Metab Dispos. 2008;36(7):1375–84.
Cleary Y, Gertz M, Morcos PN, Yu L, Youdim K, Phipps A, et al. Model-based assessments of CYP-mediated drug–drug interaction risk of alectinib: physiologically based pharmacokinetic modeling supported clinical development. Clin Pharmacol Ther. 2017.
Gillen CM, Forbush B 3rd. Functional interaction of the K-Cl cotransporter (KCC1) with the Na-K-Cl cotransporter in HEK-293 cells. Am J Phys. 1999;276(2 Pt 1):C328–36.
Mateus A, Matsson P, Artursson P. A high-throughput cell-based method to predict the unbound drug fraction in the brain. J Med Chem. 2014;57(7):3005–10.
Law V, Knox C, Djoumbou Y, Jewison T, Guo AC, Liu Y, et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42(Database issue):D1091–7.
Benet LZ, Broccatelli F, Oprea TI. BDDCS applied to over 900 drugs. AAPS J. 2011;13(4):519–47.
Vermeer LM, Isringhausen CD, Ogilvie BW, Buckley DB. Evaluation of ketoconazole and its alternative clinical CYP3A4/5 inhibitors as inhibitors of drug transporters: the in vitro effects of ketoconazole, ritonavir, clarithromycin, and itraconazole on 13 clinically-relevant drug transporters. Drug Metab Dispos. 2016;44(3):453–9.
Isoherranen N, Kunze KL, Allen KE, Nelson WL, Thummel KE. Role of itraconazole metabolites in CYP3A4 inhibition. Drug Metab Dispos. 2004;32(10):1121–31.
Higgins JW, Ke AB, Zamek-Gliszczynski MJ. Clinical CYP3A inhibitor alternatives to ketoconazole, clarithromycin and itraconazole, are not transported into the liver by hepatic organic anion transporting polypeptides and organic cation transporter 1. Drug Metab Dispos. 2014;42(11):1780–4.
Yamano K, Yamamoto K, Kotaki H, Sawada Y, Iga T. Quantitative prediction of metabolic inhibition of midazolam by itraconazole and ketoconazole in rats: implication of concentrative uptake of inhibitors into liver. Drug Metab Dispos. 1999;27(3):395–402.
Farowski F, Cornely OA, Vehreschild JJ, Hartmann P, Bauer T, Steinbach A, et al. Intracellular concentrations of posaconazole in different compartments of peripheral blood. Antimicrob Agents Chemother. 2010;54(7):2928–31.
Filppula AM, Parvizi R, Mateus A, Baranczewski P, Artursson P. Improved predictions of time-dependent drug-drug interactions by determination of cytosolic drug concentrations. Sci Rep. 2019;9(1):5850.
Kilford PJ, Gertz M, Houston JB, Galetin A. Hepatocellular binding of drugs: correction for unbound fraction in hepatocyte incubations using microsomal binding or drug lipophilicity data. Drug Metab Dispos. 2008;36(7):1194–7.
Riccardi K, Ryu S, Lin J, Yates P, Tess D, Li R, et al. Comparison of species and cell-type differences in fraction unbound of liver tissues, hepatocytes, and cell lines. Drug Metab Dispos. 2018;46(4):415–21.
Riccardi K, Lin J, Li Z, Niosi M, Ryu S, Hua W, et al. Novel method to predict in vivo liver-to-plasma Kpuu for OATP substrates using suspension hepatocytes. Drug Metab Dispos. 2017;45(5):576–80.
Rodgers T, Jones HM, Rowland M. Tissue lipids and drug distribution: dog versus rat. J Pharm Sci. 2012;101(12):4615–26.
Di L, Keefer C, Scott DO, Strelevitz TJ, Chang G, Bi YA, et al. Mechanistic insights from comparing intrinsic clearance values between human liver microsomes and hepatocytes to guide drug design. Eur J Med Chem. 2012;57:441–8.
Francis GL. Albumin and mammalian cell culture: implications for biotechnology applications. Cytotechnology. 2010;62(1):1–16.
Sun Y, Chothe PP, Sager JE, Tsao H, Moore A, Laitinen L, et al. Quantitative prediction of CYP3A4 induction: impact of measured, free, and intracellular perpetrator concentrations from human hepatocyte induction studies on drug-drug interaction predictions. Drug Metab Dispos. 2017;45(6):692–705.
Prieto Garcia L, Janzen D, Kanebratt KP, Ericsson H, Lennernas H, Lundahl A. Physiologically based pharmacokinetic model of itraconazole and two of its metabolites to improve the predictions and the mechanistic understanding of CYP3A4 drug-drug interactions. Drug Metab Dispos. 2018;46(10):1420–33.
Templeton IE, Thummel KE, Kharasch ED, Kunze KL, Hoffer C, Nelson WL, et al. Contribution of itraconazole metabolites to inhibition of CYP3A4 in vivo. Clin Pharmacol Ther. 2008;83(1):77–85.
Varma MV, Lai Y, Kimoto E, Goosen TC, El-Kattan AF, Kumar V. Mechanistic modeling to predict the transporter- and enzyme-mediated drug-drug interactions of repaglinide. Pharm Res. 2013;30(4):1188–99.
Sato M, Toshimoto K, Tomaru A, Yoshikado T, Tanaka Y, Hisaka A, et al. Physiologically based pharmacokinetic modeling of bosentan identifies the saturable hepatic uptake as a major contributor to its nonlinear pharmacokinetics. Drug Metab Dispos. 2018;46(5):740–8.
Acknowledgments
We thank Manfred Kansy for continuous support in this project. We thank Isabelle Walter, Vincent Monin, and Urs Bader for bioanalytical support. We thank Elisa Di Lenarda, Anne-Christine Cascais, and Marie-Elise Brun for support in permeability screens of RO compounds.
Funding
This study was financially supported by ARIADME, a European FP7 ITN Community’s Seventh Framework Program, grant no. 60751 (Andrea Treyer) and the Swedish research Council grants no. 2822 and 2017-01951 (Per Artursson).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(PDF 970 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Treyer, A., Ullah, M., Parrott, N. et al. Impact of Intracellular Concentrations on Metabolic Drug-Drug Interaction Studies. AAPS J 21, 77 (2019). https://doi.org/10.1208/s12248-019-0344-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-019-0344-8