
Abstract
We report on an infant who underwent gross total resection (GTR) of a posterior fossa ependymoblastoma in the second month of life followed by chemotherapy with uneventful long-term survival for 12 years. Postoperative radiotherapy has been considered obligate to have a chance for prolonged survival, but is inadvisable in infants. To our knowledge, this is the first reported long-term survival in an infant treated for ependymoblastoma.
Similar content being viewed by others

Introduction
Ependymoblastoma is an exceedingly rare and highly malignant tumor of the CNS, occurring most frequently in infancy and early childhood (Ding et al. 2014). In the 2007 WHO classification of tumors of the CNS, ependymoblastoma is classified as an embryonal tumor and is primarily considered a sub-type of primitive neuroectodermal tumors (CNS PNETs, WHO grade IV). The majority of reported cases occur in supratentorial locations. Although it is a highly malignant tumor, there is currently no standardized treatment strategy for ependymoblastoma.
Clinical results remain very poor, even after multimodal treatment with surgical resection, chemotherapy and radiatiotherapy (Ding et al. 2014; Gerber et al. 2011; Mørk and Rubinstein 1985). We report a 2-month-old boy diagnosed with posterior fossa ependymoblastoma and his uneventful survival for 12 years following gross total resection (GTR) and adjuvant chemotherapy.
Case report
This 2-month-old boy presented with a tense fontanelle, abnormal head growth, episodes of vomiting, and “sunset” gaze.
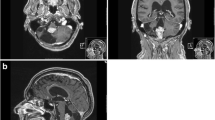
Radiographic findings An MRI revealed pronounced supratentorial hydrocephalus and a partially contrast enhancing right-sided posterior fossa tumor with downward herniation through the foramen magnum and lateral displacement of the brain stem (Fig. 1).

MRI scans disclose a partially contrast enhancing tumor to the right in the posterior fossa with marked displacement of the brain stem to the left. a sagittal, b axial and c coronal scans.
Operation Intraoperative external drainage of the cerebrospinal fluid (CSF) was established to control the intracranial pressure (ICP). A suboccipital craniotomy was performed in the mid-line. The tumor was microsurgically excised via the fourth ventricle to gross-total resection (GTR) of the tumor. The external drainage was terminated as CSF flow through the Sylvian aqueduct had been observed. Postoperative MRI scans performed in the same anesthetical procedure as the surgery confirmed the GTR (Fig. 2).

Immediate postoperative MRI scans; a sagittal, b axial, white line demonstrating gross total resection.
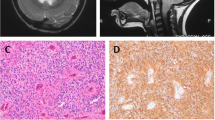
The histological examination revealed a malignant small cell tumor with several true multilayered rosettes (Fig. 3a). The rosettes were postitive for vimentin (Fig. 3b), but negative for GFAP (Fig. 3c) and synaptophysin. The tumor was also focally positive for cytokeratin. The Ki-67 labelling index was about 90% in the most cellular areas (Fig. 3d). The tumor was diagnosed as an ependymoblastoma.

A PNET-like tumor with true rosettes is seen in the HE-stained section (a). The true rosettes are positive for vimentin (b) and negative for GFAP (c). The Ki-67 labelling index is about 90% (d).
The biopsy has been re-examined by an experienced neuropathologist with the same conclusion.
Postoperative course and further treatment
Signs of persistent hydrocephalus reappeared after 3 days, and the patient underwent an endoscopic 3rd ventriculocisternostomy. This procedure did not suffice and a ventriculoperitoneal CSF-shunt had to be inserted after 2 weeks. After careful consideration the boy was given chemotherapy according to the MET-HIT 2000-BIS4 protocol during the following 40 weeks. He had several episodes with serious infections, including shunt infection. Following completion of the chemotherapy and infection controls, a new CSF shunt, now ventriculoatrial, was implanted, 14 months after the tumor resection.
Serial follow-up MRI scans have demonstrated relative normalization in the posterior fossa (Fig. 4), without any signs of recurrent neoplastic disease, as well as stable ventriculoatrial shunt function.

Late follow-up scans after 11 years. Normalisation in the posterior fossa, without signs of residual or recurrent disease.
Today, this boy is 12 years and he follows a normal school program with some assistance (5 h special teaching a week).
He has minor balance problems, but is playing soccer and he is also doing cross-country skiing.
Discussion
Ependymoblastoma is a rare subentity of PNET with ependymal differentiation and particularly poor prognosis first described by Rubinstein (1970). Most reports are of single cases, but a consecutive series of 11 children aged 1.6–5.6 years was presented by Gerber et al. (2011) They found only three cases (aged 3.4; 3.5 and 5.6 years at diagnosis) with disease free survival, and all these three were among the seven children where GTR had been achieved at primary surgery. Two of these had both postoperative radiotherapy and chemotherapy and were disease free at 12.7 and 9.4 years, the latter had developed a papillary thyroid carcinoma at the latest follow-up. The last child had complete continuous remission for 2.2 years after GTR and high dose chemotherapy (HIT 2000 with autologous stem cell transplantation).
Gerber and colleagues argue for multimodal treatment including radiotherapy to improve survival (Gerber et al. 2011). In our case, the patient was a 2-month-old infant and, in our opinion, too young for radiotherapy.
In their literature review, Ding and coworkers discuss treatment strategies for ependymoblastoma based on the 42 of 72 published cases wherefrom detailed information was available (Ding et al. 2014). They underscore the importance of GTR followed by chemotherapy. Radiation therapy should be given up-front, as this treatment modality given at a later stage does not seem to improve prognosis. They also discuss low age as a negative prognostic factor. This might be caused by biologically more aggressive tumors in infants, or by the fact that infants do not receive radiotherapy.
Conclusion
Our patient has an uneventful survival for 12 years following GTR and chemotherapy according to the HIT 2000 protocol. To our knowledge, this is the first infant with long-term survival treated for ependymoblastoma.
References
Ding D, Zhao A, Qiu B, Xing D, Guan G, Guo Z (2014) Ependymoblastoma with cystic change in a child. Case report. J Neurosurg Pediatrics 13:658–665
Gerber NU, von Hoff K, von Bueren AO, Treulieb W, Warmuth-Metz M, Pietsch T et al (2011) Outcome of 11 children with ependymoblastoma treated within the prospective HIT-trials between 1991 and 2006. J Neurooncol 102:459–469
Mørk SJ, Rubinstein LJ (1985) Ependymoblastoma. A reappraisal of a rare embryonal tumor. Cancer 55:1536–1542
Rubinstein LJ (1970) The definition of the ependymoblastoma. Arch Pathol 90:35–45
Authors’ contributions
All authors has been part of the treatment team of this patient, and has contributed in drafting the manuscript. All authors read and approved the final manuscript.
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Consent for publication Informed consent for publication of the case was given by the parents.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Due-Tønnessen, B.J., Egge, A., Lundar, T. et al. Posterior fossa ependymoblastoma diagnosed in the second month of life: uneventful 12 years survival after gross total resection followed by chemotherapy. SpringerPlus 4, 389 (2015). https://doi.org/10.1186/s40064-015-1178-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40064-015-1178-1