
Abstract
Background
The current investigation sought to explore the nature of the secondary metabolites in the algae, Laurencia pacifica.
Results
This report details the first isolation of the sesquiterpenes isoaplysin (1), isolaurenisol (2), debromoisolaurinterol (3), debromoaplysinol (4), laur-11-en-10-ol (5), 10α-hydroxyldebromoepiaplysin (6), and the previously unknown 10-bromo-3,7,11,11-tetramethylspiro[5.5]undeca-1,7-dien-3-ol (7) from the algae, Laurencia pacifica. Isoaplysin (1) and debromoaplysinol (4) showed promising levels of growth inhibition against a panel cancer-derived cell lines of colon (HT29), glioblastoma (U87, SJ-G2), breast (MCF-7), ovarian (A2780), lung (H460), skin (A431), prostate (Du145), neuroblastoma (BE2-C), pancreas (MIA), murine glioblastoma (SMA) origin with average GI50 values of 23 and 14 μM.
Conclusions
Isoaplysin (1) and debromoaplysinol (4) were up to fourfold more potent in cancer-derived cell populations than in non-tumor-derived normal cells (MCF10A). These analogues are promising candidates for anticancer drug development.

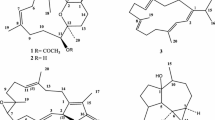
Graphical Abstract
Similar content being viewed by others


Findings
Introduction
Natural products with their high fraction sp3 content (Fsp3) represent a significant proportion of all clinical drugs [1]. Of the 1,355 new entities introduced as therapeutics between 1981 and 2010, 71% were natural products or natural product derived [2]. A high Fsp3 content imbues natural products with defined three-dimensional geometry that allows for high levels of interaction with a wide range of biological targets. A significant number of natural products adhere to the `rule of five´ and thus present high levels of drug-like character [3],[4]. Natural products also afford access to a wide range of novel chemical motifs accessing new chemical space in the drug design and development arena. This has led to the ongoing interest in accessing natural product secondary metabolites (in particular) with their high chemical diversity and biological specificity, making them a favorable source of lead compounds for drug discovery and development [5],[6].
Recently, we turned our attention to marine natural products as a potential source for new lead compounds. In this area, we have identified a small family of cytotoxic steroids from an Australian sponge Psammoclema sp. [7], and antimalarial, antialgal, antitubercular, antibacterial, antiphotosynthetic, and antifouling activity of diterpene and diterpene isonitriles from the tropical marine sponge Cymbastela hooperi[8]. In this present study, we examined the cytotoxicity of extracts obtained from Laurencia pacifica algae collected in the pacific coast of the Baja California Peninsula, Mexico.
The genus Laurencia typically inhabits the world's tropical oceans and has been responsible for approximately half of all the reported compounds from red algae. This genus is considered an important producer of halogenated sesquiterpenes, diterpenes, and acetogenins [9]-[11]. Biological activities of the Laurencia family range from antipredatory [12], antifungal [13], antibacterial [14]-[16], to anticancer [17]-[19]. Secondary metabolites reported from L. pacifica include γ-bisabolene, bromocuparane, laurinterol, debromolauriterol, isolaurinterol, aplysin, debromoaplysin, 10-bromo-α-chamigrene, prepacifenol, pacifenol, pacifidine, and kylinone [10].
Results and discussion
The algae, L. pacifica was collected from the Baja California Peninsula, Mexico. The ethanol extracts were examined for the potential presence of cytotoxic compounds. Cytotoxicity screening was conducted against a panel of cancer cell lines of colon (HT29), glioblastoma (U87, SJ-G2), breast (MCF-7), ovarian (A2780), lung (H460), skin (A431), prostate (Du145), neuroblastoma (BE2-C), pancreas (MIA), murine glioblastoma (SMA) origin, and a normal line of breast cells (MCF10A) [20]. The preliminary screening showed sufficient promise to embark on an isolation program (data not shown).
Bioassay-guided fractionation (normal phase chromatography) of the L. pacifica ethanolic extracts (see Additional file 1) resulted in the isolation of seven sesquiterpenes: isoaplysin (1) [21],[22], isolaurenisol (2) [13],[22], debromoisolaurinterol (3) [23], debromoaplysinol (4) [10],[13],[21], laur-11-en-10-ol (5) [13], 10α-hydroxyldebromoepiaplysin (6) [13] (Figure 1) and the previously unreported 10-bromo-1,7-dien-3-ol (7) (Figure 2). Sesquiterpenes 1 to 6 were identified in comparison with their spectroscopic data against literature data [21]-[23].

Chemical structures of the known sesquiterpenes isolated from Laurencia pacifica in this work. Isoaplysin (1), isolaurenisol (2), debromoisolaurinterol (3), debromoaplysinol (4), laur-11-en-10-ol (5), and 10α-hydroxyldebromoepiaplysin (6).

Important HMBC (blue arrows) and COSY (bold bonds) correlations for 7, and the chemical structure of the related 10-bromo-7,8-epoxychamigr-1-en-3-ol (8).
Sesquiterpenes 1 to 6 have been found in other Laurencia algae: L. okumurai, (1, 2, 3, 4, and 6) [21],[22], L. gracilis (2) [24],[25], L. tristicha (5) [19], and L. distichophylla (3) [26]. This work represents the first identification of these sequiterpenes in L. pacifica.
Sesquiterpene 7 was isolated in very small quantities (approximately 100 ng) from 2 kg of algae and was identified through a combination of high resolution mass spectrometry, infrared spectroscopy, and heteronuclear multiple bond correlation (HMBC), heteronuclear single quantum correlation (HSQC), correlated spectroscopy (COSY) NMR (see Additional file 1). The spectral data obtained closely matched reported spectral data of the related 10-bromo-7,8-expoxychamigr-1-en-3-ol (8) (Figure 2) [26], in which the C6 stereochemistry was determined by detailed NMR analysis and was consistent with our data [27]-[29]. The peak assignment and spectral comparison of 7 and 8 are shown in Table 1. There are three related structures with the C1-C2 double bond, one of which is supported by crystal structure data and is of the same absolute configuration as shown for 7 and 8. While these data are wholly consistent with our assignment, Suescun et al. have identified another compound with the opposite C6 configuration [29]. We thus consider our absolute configuration assignment as tentative. Notwithstanding this, the spectroscopic data is consistent with the assigned structure and represents a new sesquiterpene from L. pacifica (Figure 2).
Sequiterpenes 1 to 5 were isolated in sufficient quantities allowing direct evaluation as pure compounds against a panel of cancer and non-cancer-derived cell lines. Due to the low levels of sesquiterpenes 6 and 7, these were screened as a 1:1 mixture (both were isolated from the same extract fraction as determined by 1H NMR) [30]. Initial cytotoxicity screening was conducted at a single dose of 25 μM, and these data are presented in Table 2.
Analysis of the cytotoxicity data presented in Table 2 highlights the low level of cell death of the 12 cell lines examined on treatment with 2, 3, 5, and 6/7. Isoaplysin (1) and debromoaplysinol (4) displayed promising levels of cell death from 10% to >100% and 31% to >100% at 25 μM drug concentration, respectively. Of the other analogues, only isolaurenisol (2) displayed any growth inhibition at >20% (Du145, prostate cancer cell line). Given the activities of these three analogues, a full dose response evaluation was undertaken across our panel of carcinoma and normal cell lines [20]. These data are presented in Table 3.
As anticipated, isolaurenisol (2) displayed no noteworthy cytotoxicity returning a GI50 value > 50 μM across all cell lines examined. Both isoaplysin (1) and debromoaplysinol (4) displayed good to excellent levels of cytotoxicity. Isoaplysin (1) returned an average GI50 value of 23 μM (from 15 ± 1.2 μM to 40 ± 0.6 μM against the HT29 and U87 cell lines, respectively), while debromoaplysinol (4) returned an average GI50 value of 14 μM (from 6.8 ± 0.3 μM to 26 ± 1.7 μM against the Du145 and U87 cell lines, respectively) when screened in cancer-derived cell lines. Both compounds showed the greatest growth inhibitory effect in the prostate cancer-derived cell line Du145 with GI50 values of 12 and 6.8 μM, respectively, and the least growth inhibitory effect in the non-cancer-derived normal breast cells with GI50 values of 46 and 28 μM, respectively. Indeed, 1 and 4 were up to fourfold more potent in cancer-derived cell populations than normal cells, imbuing them with properties favorable for future development as anti-cancer agents.
Given the structural similarity of the isolated analogues (1 to 6), being direct structural homologues or biosynthetically related, the observed differences in cytotoxicity suggest that the presence of the furan moiety and positioning and nature of the pendent substituents was important for cytotoxicity. Analogues 1 and 4 are Br − OH bioisosteres, and 4 and 6 are positional isomers (C3a-OH (1) and a C10a-OH (6)). The position of the − OH moiety is clearly important with the 3α-hydroxydebromoaplysin (6) devoid of cytotoxicity, whereas debromoaplysinol (4) displays excellent levels of activity (for a potential lead compound) against the HT29 (9.1 μM), A431 (9.6 μM), and Du145 (6.8 μM) cell lines [31]. Isoaplysin (1) and debromoaplysinol (4) differ only in the presence of the C3a-Br (1) and a C3a-OH (4) moiety, and 4 displays enhanced specific and broad spectrum cytotoxicity relative to 1, suggesting that the − OH moiety enhances the cytotoxicity of this class of compounds.
Sun et al. isolated and screened the related sesquiterpenes: aplysin-9-ene, epiaplysinol, debromoepiaplysinol, aplysinol, and aplysin isolated from Laurencia tristicha in a MTT assay against lung adenocarcinoma (A549), stomach cancer (BGC-823), hepatoma (Bel 7402), colon cancer (HCT-8), and HeLa cell lines [19]. Interestingly, only debromoepiaplysinol (epi-4, this work) displayed cytotoxicity with a GI50 = 15.5 μM against HeLa cells, where as herein, debromoaplysinol (4) displays activity across our panel of 11 cancer cell lines with strong activity in the HT29 (9.1 μM), A431 (9.6 μM), and Du145 (6.8 μM) cells [19]. These findings serve to emphasize the subtle nature of drug-ligand interactions and the role of stereochemistry in eliciting biological activity, c.f. 1, 4, and 6.
Conclusions
Herein, we have identified sesquiterpenes 1 to 6 for the first time in L. pacifica and isolated a new sesquiterpene and 10-bromo-1,7-dien-3-ol (7). Screening of these analogues against 11 cancer cell lines revealed modest to good levels of cytotoxicity for 1 and 4, with up to fourfold selectivity towards cancer-derived cell populations compared with normal cells. Given the low molecular weights and high Fsp3 content, the structure activity data elucidated in this small subset of analogues, we believe that 1 and 4 represent excellent leads for the development of selective and potent anticancer agents [31].
Experimental
General experimental
Solvents. Solvents used for TLC, speedy column, and centrifugal chromatography were of bulk quality and were distilled from glass prior to use. In the case of HPLC, all of the solvents were HPLC grade and were filtered and degassed prior to their use. The solvent referred as LP (light petroleum), is a mixture of different alkanes with a boiling point 60°C to 80°C.
Collection of the Mexican algae
L. pacifica algae were collected on the coast of the Baja California Peninsula, Mexico. The algae was cleaned of epiphytes, rinsed with fresh water, and dried in the sun at the collection site. The specimens were stored at −20°C. A voucher specimen of L. pacifica was preserved on location in 5% formaldehyde and deposited in a private collection at the Algal Laboratory in the Interdisciplinary Center of Marine Sciences (CICIMAR), La Paz, B.C.S., Mexico, for taxonomical identification and future reference. Subsequently, in the laboratory, 10 g of dry algae was roughly torn or cut to small pieces and then ground with a mortar and pestle. The powdered algae was then submerged in 250 mL of ethanol. The mixture was left for 48 h at 25°C to 35°C. Afterwards, the mixture was filtered and the residual algal tissue was extracted again under the same conditions. Both filtered extracts were combined and concentrated to dryness under reduced pressure at 40°C to obtain ca 30 mg of extract. These extracts were used for biological screening.
Extracts of L. pacifica and its fractionation
Crude extract of L. pacifica 2 kg of algae was reduced to small pieces of ca as before, and then submerged in 1 L of ethanol. The resulting mixture was left for 48 h at 25°C to 35°C. Afterwards, the mixture was filtered and the residual algal tissue was extracted again under the same conditions. Both filtered extracts were combined and concentrated to dryness under reduced pressure at 40°C to obtain 2.2 g of extract. Fractionation of the crude extract was commenced with a speedy column resulting in 18 fractions [32]. All fractions were tested in the colorimetric assay; active fractions were then fractionated in normal phase HPLC until isolation of a pure compound.
NMR
Proton and 13C NMR spectra were recorded on a Bruker Ascend 400 or Bruker Ascend 600 (Madison, WI, USA). All NMR spectra were recorded as CDCl3 solutions; the solvent signal was used as internal standard for chemical shifts (13C δ 77.0 ppm, and 1H δ 7.24 ppm for the residual CHCl3 proton). All spectra, including HSQC, HMBC, distortionless enhancement by polarization transfer (DEPT135), distortionless enhancement by polarization transfer with retention of quaternaries (DEPTQ135), and (homonuclear) COSY-utilized standard Bruker pulse programs.
Cell culture and stock solutions
Stock solutions were prepared as follows and stored at −20°C: drugs were stored as 20 mM solutions in DMSO. All cell lines were cultured at 37°C, under 5% CO2 in air. All cancer-derived cells lines were maintained in Dulbecco's modified Eagle's medium (Trace Biosciences, Sydney, Australia) supplemented with 10% fetal bovine serum, 10 mM sodium bicarbonate, penicillin (100 IU/mL), streptomycin (100 μg/mL), and glutamine (4 mM). The non-cancer-derived breast cell line MCF10A was maintained in Dulbecco's modified Eagle's medium and Ham's F12 medium (1:1, Trace Biosciences, Sydney, Australia) supplemented with 5% heat inactivated horse serum, HEPES (20 mM), penicillin (100 IU/ml), streptomycin (100 μg/mL), glutamine (2 mM), epidermal growth factor (20 ng/ml), hydrocortisone (500 mg/ml), cholera toxin (100 ng/ml), and insulin (10 μg/mL).
In vitro growth inhibition assay
Cells in logarithmic growth were transferred to 96-well plates. Cytotoxicity was determined by plating cells in duplicate in 100 mL medium at a density of 2,500 to 4,000 cells/well. On day 0, (24 h after plating) when the cells were in logarithmic growth, 100 μL medium with or without the test agent was added to each well. After 72 h, drug exposure growth inhibitory effects were evaluated using the MTT (3-[4,5-dimethyltiazol-2-yl]-2,5-diphenyl-tetrazolium bromide) assay and absorbance read at 540 nm. Percentage growth inhibition was determined at a fixed drug concentration of 25 μM. A value of 100% is indicative of total cell growth inhibition. Those analogues showing appreciable percentage growth inhibition underwent further dose response analysis allowing for the calculation of a GI50 value. This value is the drug concentration at which cell growth is 50% inhibited based on the difference between the optical density values on day 0 and those at the end of drug exposure [20].
Isoaplysin: (3S,3aS,8bS)-3a-(Bromomethyl)-3,6,8b-trimethyl-2,3,3a,8b-tetrahydro-1H-benzo[b]cyclopenta[d]furan ( 1 )
Isolated as a white powder: 1.5 mg; [α] = −5.3° (c 0.001, CH3OH); IRνmax 2,927 (C−H), 2,858, 1,620 (C = C), 1,593 (C = C), 1,499, 1,453 (C = C), 1,423, 1,376 (C−H), 1,268 (C−O), 1,135, 946 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.88 (d, J = 7.6 Hz, 1H, H-5), 6.66 (dd, J = 7.5, 0.7 Hz, 1H, H-4), 6.58 (s, 1H, H-2), 3.66, 3.56 (ABq, JAB = 11.1 Hz, 2H, H-12), 2.27 (s, 3H, H-15), 2.21 to 2.12 (m, 1H, H-10), 1.90 to 1.83 (m, 1H, H-8a), 1.70 to 1.60 (m, 2H, H-8b, H-9a), 1.50 (s, 3H, H-14), 1.20 to 1.13 (m, 1H, H-9b), 1.10 (d, J = 6.7 Hz, 3H, H-13). 13C NMR (101 MHz, CDCl3) δ 158.8 (q C, C-1), 138.3 (q C, C-3), 133.0 (q C, C-6), 122.2 (CH, C-5), 121.4 (CH, C-4), 109.3 (CH, C-2), 97.2 (q C, C-11), 55.5 (q C, C-7), 43.7 (CH, C-10), 42.6 (CH2, C-8), 34.6 (CH2, C-12), 31.5 (CH2, C-9), 22.9 (CH3, C-14), 21.5 (CH3, C-15), 13.8 (CH3, C-13).
Isolaurenisol: 2-[3-(Bromomethylene)-1,2-dimethylcyclopentyl]-5-methylphenol ( 2 )
Isolated as a white solid: 2.5 mg; [α] = −7.2° (c 0.0025, CH3OH); IRνmax 3,513 (O-H), 2,959 (C-H), 2,929, 2,870, 1,616 (C = C), 1,576 (C = C), 1,514, 1,454, 1,412, 1,294 (C-H), 1,254 (C-O), 1,186, 1,123, 809, 787, 653 (C-Br) cm−1; 1H NMR (400 MHz, CDCl3) δ 7.15 (d, J = 7.9 Hz, 1H, H-5), 6.68 (dd, J = 7.9, 1.0 Hz, 1H, H-4), 6.57 (d, J = 1.1 Hz, 1H, H-2), 5.99 (d, J = 2.0 Hz, 1H, H-13), 5.06 (s, 1H, OH), 3.05 to 2.95 (m, 1H, H-11), 2.56 (dt, J = 12.9, 7.2 Hz, 1H, H-8a), 2.25 (s, 3H, H-15), 2.07 to 1.96 (m, 1H, H-9a), 1.64 to 1.57 (m, 1H, H-8b), 1.45 (s, 3H, H-14), 1.42 to 1.36 (m, 1H, H-9b), 1.22 (d, J = 7.2 Hz, 3H, H-12). 13C NMR (101 MHz, CDCl3) δ 160.2 (q C, C-10), 153.3 (q C, C-1), 138.0 (q C, C-3), 128.7 (q C, C-6), 128.1 (CH, C-5), 121.3 (CH, C-4), 118.2 (CH, C-2), 101.3 (CH, C-13), 52.0 (q C, C-7), 39.2 (CH, C-11), 39.2 (CH2, C-8), 31.0 (CH2, C-9), 26.8 (CH3, H-14), 20.7 (CH3, C-15), 19.1 (CH3, C-12).
Debromoisolaurinterol: [(1 R ,3 S )-1,3-dimethyl-2-methylenecyclopentyl]-5-methyl-2-phenol ( 3 )
Isolated as a colorless oil: ca 1 mg; IRνmax 3,458 (O-H), 3,055 (C-H), 2,984 (C-H), 1,641 (C = C), 1,581 (C = C), 1,113 (C-H) cm-1; 1H NMR (400 MHz, CDCl3) δ 7.21 (d, J = 8.0 Hz, 1H, H-5), 6.71 (dd, J = 8.0, 1.1 Hz, 1H, H-4), 6.65 (d, J = 1.4 Hz, 1H, H-2), 5.54 (s, 1H, OH), 5.08 (d, J = 2.1 Hz, 1H, H-12a), 4.93 (d, J = 2.4 Hz, 1H, H-12b), 2.88 to 2.78 (m, 1H, H-10), 2.26 (s, 3H, H-15), 2.25 to 2.18 (m, 1H, H-8a), 2.07 to 1.98 (m, 1H, H-9a), 1.60 to 1.55 (m, 1H, H-8b, obscured), 1.46 (s, 3H, H-14), 1.41 to 1.36 (m, 1H, H-9b), 1.19 (d, J = 7.0 Hz, 3H, H-13).
Debromoaplysinol: 3a-methanol, 1,2,3,8b-Tetrahydro-3,6,8b-trimethylcyclopenta-3 H -[ b ]benzofuran ( 4 )
Isolated as a colorless oil: ca 1 mg; [α] = 0° (c 0.001, CH3OH); IRνmax 3,425 (O-H), 2,940, 2,871, 1,588 (C = C), 1,499, 1,044 (C-O), 860 cm-1; 1H NMR (400 MHz, CDCl3) δ 6.90 (d, J = 7.6 Hz, 1H, H-5), 6.66 (dd, J = 7.5, 0.7 Hz, 1H, H-4), 6.57 (s, 1H, H-2), 3.84 (dABq, J = 12.4(AB), 4.3 Hz, 1H, H-12a), 3.71 (dABq, J = 12.4(AB), 8.6 Hz, 1H, H-12b), 2.28 (s, 3H, H-15), 1.88 to 1.78 (m, 2H, H-10, H-8a), 1.72 (dd, J = 8.6, 4.3 Hz, 1H, OH), 1.66 to 1.60 (m, 2H, H-8b, H-9a), 1.46 (s, 3H, H-14), 1.16 to 1.11 (m, 1H, H-9b), 1.08 (d, J = 6.8 Hz, 3H, H-13). 13C NMR (101 MHz, CDCl3) δ 159.1 (q C, C-1), 138.3 (q C, C-3), 133.3 (q C, C-6), 122.2 (CH, C-5), 121.2 (CH, C-4), 109.0 (CH, C-2), 99.6 (q C, C-11), 63.9 (CH2, C-12), 54.2 (q C, C-7), 42.3 (CH, C-10), 42.3 (CH2, C-8), 31.5 (CH2, C-9), 22.9 (CH3, C-14), 21.2 (CH3, C-51), 13.7 (CH3, C-13).
Laur-11-en-10-ol. 3-(4′-Methylphenyl)-1,3,dimethyl-2-methylidenecyclopentanol ( 5 )
Isolated as a colorless oil: ca 1 mg; 1H NMR (600 MHz, CDCl3) δ 7.21 (d, J = 7.9 Hz, 2H, H-1, H-5), 7.07 (d, J = 8.1 Hz, 2H, H-2, H-4), 5.44 (s, 1H, H-12a), 4.99 (s, 1H, H-12b), 3.47 (d, J = 5.9 Hz, 1H, OH), 2.29 (s, 3H, H-15), 2.08 to 2.04 (m, 1H, H-8a), 1.98 to 1.92 (m, 1H, H-8b), 1.82 to 1.77 (m, 1H, H-9a), 1.67 to 1.62 (m, 1H, H-9b, obscured), 1.48 (s, 3H, H-14), 1.36 (s, 3H, H-13). 13C NMR (151 MHz, CDCl3) δ 166.4 (q C, C-11), 145.4 (q C, C-6), 135.3 (q C, C-3), 129.0 (2 ± CH, C-2, C-4), 126.3 (2 ± CH, C-1, C-5), 108.6 (CH2, C-12), 79.3 (q C, C-10), 50.3 (q C, C-7), 39.5 (CH2, C-8), 39.2 (CH2, C-9), 30.8 (CH3, C-14), 28.4 (CH3, C-13), 21.2 (CH3, C-15).
10α-hydroxyldebromoepiaplysin. (−)-2,3,3a,8b-Tetrahydro-3-hydroxy-3,3a,6,8b-tetramethyl-1H-benzocyclopentafuran ( 6 )
Isolated as a colorless oil: ca 1 mg; 1H NMR (600 MHz, CDCl3) δ 6.92 (d, J = 7.6 Hz, 1H, H-5), 6.66 (d, J = 7.4 Hz, 1H, H-4), 6.49 (s, 1H, H-2), 5.35 (bs, 1H, OH), 2.26 (s, 3H, H-15), 2.01 to 1.96 (m, 1H, H-8a, obscured), 1.76 to 1.71 (m, 1H, H-8b, obscured), 1.61 to 1.57 (m, 2H, H-9, obscured), 1.39 (s, 3H, H-13), 1.38 (s, 3H, H-14), 1.28 (s, 3H, H-12). 13C NMR (151 MHz, CDCl3) δ 158.0 (q C, C-1), 138.1 (q C, C-3), 133.5 (q C, C-6), 122.5 (CH, C-5), 121.0 (CH, C-4), 109.6 (CH, C-2), 100.5 (q C, C-11), 82.9 (q C, C-10), 53.6 (q C, C-7), 40.8 (CH2, C-8), 37.0 (CH2, C-9), 23.4 (CH3, C-14), 22.2 (CH3, C-13), 21.1 (CH3, C-15), 14.8 (CH3, C-12).
10-Bromo-3,7,11,11-tetramethylspiro[5.5]undeca-1,7-dien-3-ol ( 7 )
Isolated as a colorless oil: ca 0.1 mg; 1H NMR (600 MHz, CDCl3) δ 5.84 (d, J = 10.4 Hz, 1H, H-4), 5.53 (d, J = 10.5 Hz, 1H, H-5), 5.22 (s, 1H, H-8), 4.63 (dd, J = 10.8, 6.3 Hz, 1H, H-10), 2.65 to 2.59 (m, 1H, H-9a), 2.57 to 2.50 (m, 1H, H-9b), 2.02 to 1.96 (m, 2H, H-1), 1.82 to 1.77 (m, 1H, H-2a), 1.76 to 1.71 (m, 1H, H-2b, obscured), 1.55 (s, 3H, H-14, obscured), 1.29 (s, 3H, H-15), 1.10 (s, 3H, H-13), 1.01 (s, 3H, H-12). 13C NMR (151 MHz, CDCl3) δ 139.0 (q C, C-7), 136.4 (CH, C-4), 131.1 (CH, C-5), 120.7 (CH, C-8), 67.5 (q C, C-3), 61.3 (CH, C-10), 47.1 (q C, C-6), 41.5 (q C, C-11), 36.1 (CH2, C-2), 35.8 (CH2, C-9), 28.6 (CH3, C-15), 28.2 (CH2, C-1), 26.1 (CH3, C-13), 21.7 (CH3, C-14), 17.8 (CH3, C-12).
Additional file
References
López-Vallejo F, Giulianotti MA, Houghten RA, Medina-Franco JL: Expanding the medicinally relevant chemical space with compound libraries. Drug Dis Today 2012, 17: 718–726. 10.1016/j.drudis.2012.04.001
Newman DJ, Cragg GM: Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod 2012, 75: 311–335. 10.1021/np200906s
Harvey AL: Natural products in drug discovery. Drug Dis Today 2008, 13: 894–901. 10.1016/j.drudis.2008.07.004
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ: Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 2001, 46: 3–26. 10.1016/S0169-409X(00)00129-0
Koehn FE, Carter GT: The evolving role of natural products in drug discovery. Nat Rev Drug Dis 2005, 4: 206–220. 10.1038/nrd1657
Newman DJ: Natural products as leads to potential drugs: an old process or the new hope for drug discovery? J Med Chem 2008, 51: 2589–2599. 10.1021/jm0704090
Holland IP, McCluskey A, Sakoff JA, Chau N, Robinson PJ, Motti CA, Wright AD, Van Altena IA: New cytotoxic steroids from an Australian sponge Psammoclema sp. J Nat Prod 2009, 72: 102–106. 10.1021/np800688f
Wright AD, McCluskey A, Robertson MJ, MacGregor K, Gordon CP, Guenther J: Anti-malarial, anti-algal, anti-tubercular, anti-bacterial, anti-photosynthetic, and anti-fouling activity of diterpene and diterpene isonitriles from the tropical sponge Cymbastela hooperi . Org Biomol Chem 2011, 9: 400–407. 10.1039/c0ob00326c
Maschek J, Baker B: The chemistry of algal secondary metabolism. In Algal Chemical Ecology. Edited by: Maschek J, Baker B. Springer, Germany; 2008:1–20. Chapter 1 10.1007/978-3-540-74181-7_1
Eickson KL: Constituents of Laurencia. In Marine Natural Products: Chemical and Biological Perspectives. Edited by: Scheuer PJ. Academic, USA; 1983:131–257. Chapter 4 10.1016/B978-0-12-624005-4.50011-2
Ji N-Y, Xiao-Ming Li X-M, Lia K, Bin-Gui Wang B-G: Halogenated sesquiterpenes from the marine red alga Laurencia saitoi ( Rhodomelaceae ). Helvet Chim Acta 2009, 92: 1873–1897. 10.1002/hlca.200900073
Hay ME: Marine chemical ecology: chemical signals and cues structure marine populations, communities, and ecosystems. Annu Rev Mar Sci 2009, 1: 193–212. 10.1146/annurev.marine.010908.163708
Shui-Chun M, Yue-Wei G: Sesquiterpenes from Chinese red alga Laurencia okamurai . Chin J Nat Med 2010, 8: 321–325.
Crews P, Selover SJ: Comparison the sesquiterpenes from the seaweed Laurencia pacifica and its epiphyte Erythrocystis saccata . Phytochemistry 1986, 25: 1847–1852. 10.1016/S0031-9422(00)81160-7
Sims JJ, Fenical W, Wing RM, Radlick P: Marine natural products. IV. Prepacifenol, a halogenated epoxy sesquiterpene and precursor to pacifenol from the red alga, Laurencia filiformis. J Am Chem Soc 1973, 95: 972–972. 10.1021/ja00784a083
Sims JJ, Donnell MS, Leary JV, Lacy GH: Antimicrobial agents from marine algae. Antimicrob Agents Chemother 1975, 7: 320–321. 10.1128/AAC.7.3.320
Dembitsky VM, Gloriozova TA, Poroikov VV: Natural peroxy anticancer agents. Mini-Rev Med Chem 2007, 7: 571–589. 10.2174/138955707780859396
Sun J, Shi D, Ma M, Li S, Wang S, Han L, Yang Y, Fan X, Shi J, He L: Sesquiterpenes from the red alga Laurencia tristicha. J Nat Prod 2005, 68: 915–919. 10.1021/np050096g
Sun J, Shi D, Li S, Wang S, Han L, Fan X, Yang Y, Shi J: Chemical constituents of the red alga Laurencia triticha . J Asian Nat Prod Res 2007, 9: 725–734. 10.1080/10286020601103338
Thaqi A, Scott JL, Gilbert J, Sakoff JA, McCluskey A: Synthesis and biological activity of Δ-5,6-norcantharimides: importance of the 5,6-bridge. Eur J Med Chem 2010, 45: 1717–1723. 10.1016/j.ejmech.2010.01.004
Suzuki M, Kurosawa E: New aromatic sesquiterpenoids from the red alga Laurencia Okamurai yamada. Tetrahedron Lett 1978, 28: 2503–2506.
Suziki M, Kurata K, Kurosawa E: The structure of isoaplysin, a brominated rearranged cuparane-type sesquiterpenoid from the red alga Laurencia okamurai Yamada. Bull Chem Soc Jpn 1986, 59: 3981–3982. 10.1246/bcsj.59.3981
Harrowven DC, Lucas MC, Howes PD: The synthesis of a natural product family: from debromoisoluaurinterol to the aplysins. Tetrahedron 2001, 57: 791–804. 10.1016/S0040-4020(00)01055-3
Blunt JW, Lake RJ, Munro MHG: Sesquiterpenes from the marine red alga Laurencia distichophylla . Phytochemistry 1984, 23: 1951–1954. 10.1016/S0031-9422(00)84948-1
Yamada K, Yazawa H, Uemura D, Toda M, Hirata Y: Total synthesis of (±)aplysin and (±)debromoaplysin. Tetrahedron 1969, 25: 3509–3520. 10.1016/S0040-4020(01)82886-6
Li X-D, Miao F-P, Li K, Ji N-Y: Sesquiterpenes and acetogenins from the marine red alga Laurencia okamurai . Fitoterapia 2012, 83: 518–522. 10.1016/j.fitote.2011.12.018
Davyt D, Fernandez R, Suescun L, Mombrú AW, Saldaña J, Domínguez L, Coll J, Fujii MT, Manta E: New sesquiterpene derivatives from the red alga Laurencia scoparia : isolation, structure determination, and anthelmintic activity. J Nat Prod 2001, 64: 1552–1555. 10.1021/np0102307
König GM, Wright AD: Laurencia rigida : chemical investigation of its antifouling dichloromethane extract. J Nat Prod 1997, 60: 967–970. 10.1021/np970181r
Suescun L, Mombrú AW, Mariezcurrena RA, Davyt D, Fernandez R, Manta E: Two natural products from the alga Laurencia scoparia . Acta Cryst Section C 2001, C57: 286–288. 10.1107/S0108270100017935
Kelman D, Wright AD: The importance of 1H-nuclear magnetic resonance spectroscopy for reference standard validation in analytical sciences. PLoS One 2012, 7: e42061. 10.1371/journal.pone.0042061
Leeson PD, Springthorpe B: The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev Drug Dis 2007, 6: 881–890. 10.1038/nrd2445
Harwood LM: Dry-column flash chromatography. Aldrichimica Acta 1985, 18: 25–26.
Acknowledgements
DZAP gratefully acknowledges scholarship support from the Mexican government (Nacional Councli of Science and Technology (Consejo Nacional de Ciencia y Technologia, CONCYT)) and the University of Newcastle.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Zaleta-Pinet, D.A., Holland, I.P., Muñoz-Ochoa, M. et al. Cytotoxic compounds from Laurencia pacifica. Org Med Chem Lett 4, 8 (2014). https://doi.org/10.1186/s13588-014-0008-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13588-014-0008-8