
Abstract
Background
Mammalian hairs are one of the most ubiquitous types of trace evidence collected in the course of forensic investigations. However, hairs that are naturally shed or that lack roots are problematic substrates for DNA profiling; these hair types often contain insufficient nuclear DNA to yield short tandem repeat (STR) profiles. Whilst there have been a number of initial investigations evaluating the value of metagenomics analyses for forensic applications (e.g. examination of computer keyboards), there have been no metagenomic evaluations of human hairs—a substrate commonly encountered during forensic practice. This present study attempts to address this forensic capability gap, by conducting a qualitative assessment into the applicability of metagenomic analyses of human scalp and pubic hair.
Results
Forty-two DNA extracts obtained from human scalp and pubic hairs generated a total of 79,766 reads, yielding 39,814 reads post control and abundance filtering. The results revealed the presence of unique combinations of microbial taxa that can enable discrimination between individuals and signature taxa indigenous to female pubic hairs. Microbial data from a single co-habiting couple added an extra dimension to the study by suggesting that metagenomic analyses might be of evidentiary value in sexual assault cases when other associative evidence is not present.
Conclusions
Of all the data generated in this study, the next-generation sequencing (NGS) data generated from pubic hair held the most potential for forensic applications. Metagenomic analyses of human hairs may provide independent data to augment other forensic results and possibly provide association between victims of sexual assault and offender when other associative evidence is absent. Based on results garnered in the present study, we believe that with further development, bacterial profiling of hair will become a valuable addition to the forensic toolkit.
Similar content being viewed by others
Background
Over the last decade, the development of bacterial culture-independent approaches (metagenomics), based on 16S rRNA genes (hereafter referred to as 16S), sequences has become the cornerstone of microbial ecology [1]. The advent of next-generation sequencing (NGS) technologies and platforms capable of generating millions of sequences per sample facilitated assessments of microbial communities between body sites and individuals [2],[3]. The increased sequencing power stimulated the development of robust computational programmes capable of processing large, complex sequencing data sets [4] and enabled phylogenetic analyses of human and environmental genomes [5],[6].
Studies on the human microbiome (the collective genomes present in the human body) suggest that there are significant differences in bacterial composition not only between different body sites but also between individuals [3],[5],[7]. The potential that individuals may harbour unique bacterial species is of significance to forensic investigations.
For centuries, associative hair evidence relied solely on comparative microscopy based on qualitative features such as colour and pigmentation [8]-[10]. The advent of PCR in the mid-1980s initiated a paradigm shift in the forensic examination of hairs. For the first time, DNA profiles could complement qualitative microscopical observations [11]. However, the success of the highly discriminatory short tandem repeat (STR) profiling is dependent on hairs bearing anagen roots (actively growing hairs) that are rich in nuclear DNA (nuDNA) and, to a lesser extent, hairs that are in the quiescent (catagen) growth phase [12]. However, the majority of hairs recovered in forensic investigations are shed hairs (i.e. those in their telogen phase); these hairs have ceased to grow and contain little or no nuDNA [13]. STR profiling of these hair roots typically yields trace amounts of, often degraded, human DNA and can require the use of low-template DNA strategies and the complications that accompany such approaches [14]. In these instances, mitochondrial DNA (mtDNA) analysis is routinely conducted. However, due to its common matrilineal inheritance and haploid nature, mtDNA typing yields modest exclusionary capability, which lacks the statistical power afforded by STR profiling [15]. However, low yields of human nuDNA from forensic hair samples does not equate to the absence of other sources of DNA that could assist in the individualisation of hair. Indeed, metagenomic analyses of hairs unsuitable for nuDNA profiling may provide a microbial fingerprint to augment other forensic results such as mtDNA analyses. This would not involve extra or additional extraction procedures, as DNA isolation procedures for human DNA will also ‘collect’ microbial DNA.
Conventional forensic hair examination, using either morphological or molecular techniques, is contingent upon the deposition and recovery of hairs; however, despite Locard’s adage that ‘every contact leaves a trace’ [16], this may not always be the case. Research in relation to the transfer of pubic hairs in forensic investigations involving sexual assault cases discovered limited transfer (4%) of male pubic hair to female genital area during sexual intercourse (SI) [17]. In addition, the present study demonstrated that no female pubic hair transfer to male genital area took place.
The utility of metagenomic analyses for forensic applications has been explored since the inception of NGS; for example, Fierer et al. [18] conducted preliminary work to explore the potential to link individuals to computer keyboards and mice on the basis of transfer of skin bacteria. However, one of the most ubiquitous of evidence types—human hair—has yet to be evaluated in the context of forensic metagenomics. To the best of our knowledge, this present study is the first to qualitatively assess the viability of metagenomic analyses of hairs in a forensic context. The three aims of the research reported here were to assess:
-
1)
Whether human scalp and pubic hairs can be differentiated on the basis of their 16S microbial composition
-
2)
Whether individuals can be differentiated on the basis of microbial taxa colonising scalp and pubic hairs
-
3)
Whether bacterial 16S profiles on hair shafts are stable over time
Overall, the objective of this initial study was to establish whether further development of the technique is warranted.
Methods
Sample collection
Bacterial communities, associated with human scalp and pubic hair, were surveyed using a multiplex barcoded sequencing approach from seven healthy Caucasian individuals of both sexes (two of whom were in a de facto relationship), ranging in age from 23 to 53 years old. The health status of each volunteer was self-reported with each individual stating that antibiotics were not taken at least 8 months prior to the collection of hairs used in the study. Each individual self-collected a number of hairs cut from the scalp and pubic areas, at three time points, initial collection in addition to 2 and 5 months thereafter, referred to as T0, T2 and T5 respectively. Replication is important in NGS amplicon sequencing workflows—due to the investigative nature of this study and limited availability of resources, we selected to investigate multiple time points (temporal replicates) in lieu of multiple extractions at each time points (sampling replicates).
Each volunteer was provided with a hair collection kit consisting of labelled clip-seal plastic bags, sterilised scissors, ethanol wipe, latex disposable gloves and disposable forceps. Hairs were cut close to the skin and from approximately the same area at each time point; volunteers were asked to clean the scissors with ethanol wipes between sampling to avoid contamination. The rationale of severing the hairs close to the skin, rather than plucking the hairs was to ensure the bacterial taxa identified were more likely to originate from the hair shaft rather than the skin. Hairs taken from the head were labelled as female scalp hair (FSH) or male scalp hair (MSH); similarly, female and male pubic hairs were marked female pubic hair (FPH) or male pubic hair (MPH). Once sampled, these hairs were placed in separate labelled clip-seal plastic bags and stored at room temperature after being catalogued. Hairs were sampled and processed within 24 h of collection, and unused hair samples were returned to their original packaging and stored at room temperature; these hairs were not further sampled or examined. The effect of storage and storage conditions on bacteria was not in the scope of this present study; however Lauber et al. [19] investigated the effects of storage conditions on bacteria and concluded that bacterial community composition is unaffected in the short-term.
Each volunteer was made aware of the nature of the study and gave written, informed consent. Information regarding the sexual habits or orientations of the volunteers was not sought. The project was approved by, and conducted in accordance with, Murdoch University Human Research Ethics Committee Policies and Guidelines (Project Number 2011/139).
DNA extraction and quantification
Three hairs from each body area were cut into approximately 1 cm lengths and placed into 1.5 ml Eppendorf tubes. The contents of each tube were digested overnight using 1 ml of hair digest buffer containing: 10 mM Tris pH 8 (Sigma, St. Louis, Mo, USA), 10 mM NaCl (Sigma), 5 mM CaCl2, (Sigma), 2.5 mM EDTA pH 8 (Invitrogen, Carlsbad, CA, USA), 1 mg/ml ProK (Amresco, Solon, OH, USA), 40 mM DTT (Thermo Fisher Scientific, Waltham, MA, USA) 2% SDS (Invitrogen) and milliQ water (Sigma) to make the remaining volume. The samples, including extraction and laboratory environmental controls, were secured on rotating arms (to ensure total immersion) and digested overnight in a 55°C oven.
All samples were then centrifuged for 2 min at 13,000 rpm. To concentrate the DNA, a total of 600 μl of supernatant was transferred to Vivaspin ultrafiltration spin columns with a 30,000 MW cutoff (Sartorius Stedim Biotech, Göttingen, Germany) and centrifuged at 30,000 rpm to leave 50–100 μl of supernatant. Concentrated supernatant was subsequently combined with five volumes of PB buffer (Qiagen, Valencia, CA, USA) and transferred to a Qiagen silica spin column and centrifuged at 13,000 rpm for 1 min. Two wash steps followed (Qiagen AWI buffer and AWII buffer) prior to elution of DNA from the spin column with 60 μl of 10 mM Tris-Cl pH 8 buffer. The DNA extracts were subsequently quantified via real-time quantitative polymerase chain reaction (qPCR; Applied Biosystems StepOne, Foster City, CA, USA) using SYBR green and the bacterial 16S F515/Bact 16S (V4 loop)_R806 primers (Table 1).
Extracts were analysed using qPCR for neat extracts in addition to 1/10 and 1/100 dilutions, in order to determine if extractions were successful and to identify samples with low-template DNA (defined as those with CT values >32). The possible presence of PCR inhibitors was also determined by qPCR. The 16S qPCR assay was conducted in 25 μl reactions using a 2X ABI Power SYBR master mix (Applied Biosystems) together with 2 μl of extracted DNA with primer concentration at 0.4 μM (IDT) cycled for 95°C for 5 min followed by 50 cycles of 95°C for 30 s, 55°C for 30 s, 72°C for 45 s, with 1°C melt step and a 10-min final extension at 72°C. The optimal DNA concentration free of inhibition was used for all subsequent analyses. Each hair sample bacterial extract had CT values less than 32 PCR cycles indicating the presence of sufficient 16S template copy number for robust NGS amplicon sequencing.
Fusion-tagged 16S V4 Amplicon generation
Bacterial F515 and R806 (Table 1) 16S primers (targeting the V4 region) used in the initial qPCR extract screen, giving a size variable product minus primers of ~250 base pairs, were modified into fusion primers for the generation of amplicon products for subsequent sequencing. Each fusion primer consisted of a GS FLX Titanium (Lib-A) adapter A or B on the 5′ end followed by a unique 6 bp multiplex identifier (MID) tag and the template specific forward or reverse primer at the 3′ end of the primer [22] giving a final size variable product of ~350 bp including primers and additions. A single-step, uniquely tagged fusion PCR approach was employed to minimise the contamination associated with the multiple PCR steps used in NGS workflows [20].
For each time point, each extract was assigned a unique 6 bp MID-tagged fusion primer in preparation for amplicon sequencing. MID-tagged amplicons were generated in triplicate (i.e. PCR replicates) in 25 μL reactions containing: 1X PCR Gold Buffer (Applied Biosystems), 2.5 mM MgCl2 (Applied Biosystems), 0.1 mg/ml BSA (Fisher Biotech, WA, Australia), 0.25 mM of each dNTP (Astral Scientific, NSW, Australia), 0.4 μM of forward and reverse primer (IDT), 0.2 μL1 unit of Taq DNA polymerase (AmpliTaq Gold™), 1:80,000 (final concentration) of SYBR Green ‘gel-stain’ (Life Technologies, S7563, Carlsbad, CA, USA) and DNA extract. The same processes were performed on PCR negative/reagent controls for each PCR plate run, pre- and post addition of DNA extracts.
All PCR amplicons were purified using the Agencourt AMPure XP™ Bead PCR Purification protocol (Beckman Coulter Genomics, Danvers, MA, USA). Solely for the purpose of sequencing coverage, purified amplicons were electrophoresed on 2% agarose gel to obtain roughly equimolar ratios of each sample. Where extraction/environmental controls or PCR negative/reagent controls showed positive qPCR results, these were also pooled, purified and sequenced. The final pooled library was quantified using qPCR to determine the appropriate volume of library to use for emulsion PCR (emPCR) prior to amplicon sequencing on the GS Junior™ (described in Murray et al. [21], using reaction conditions in Murray et al. [23]). All emPCR, bead recovery and amplicon sequencing procedures were carried out according to Roche GS Junior™ protocols for amplicon sequencing (Lib A).
Bioinformatic analysis
Amplicon sequence reads obtained from the GS-Junior™ (hereafter referred to as sequences) were sorted into batches based on MID tags assigned to each extract allowing for no mismatch in MID tag DNA sequences. Additionally, template-specific 16S bacterial primer sequences were annotated and trimmed from all sequences allowing for no mismatch in base composition or primer sequence length. Sequences that failed to meet these criteria were discarded. The aforementioned steps were conducted using Geneious™ v7.0.6 [24].
Once batched and trimmed, sequence FASTA files were imported into QIIME V1.8.0 [25] and merged into a single FASTA file. Chimeric sequences were identified and removed on a per individual sample basis using the usearch61 [26] de novo method passing --split_by_sampleid. Following this, operational taxonomic units (OTUs) were identified using an open reference OTU picking method using usearch61 with a 97% clustering identity, using the most abundant sequence within each OTU as the representative sequence and the Greengenes 13.8 database release [27]. Representative sequences for each OTU were aligned using PyNAST [28] against the Greengenes 13.8 pre-aligned database, the alignment filtered and phylogeny built using FastTree [29] in QIIME. Additionally, any OTUs found within the control samples of specific time points (i.e. T0, T2, T5) were removed from samples contained within the respective time point. Following the removal of control OTUs, each individual sample was filtered to remove low abundant OTU clusters. In each case, all singleton OTUs were discarded and any OTU whose abundance was below 0.2%, an estimated error rate associated with 454 sequencing [30], of the total number of filtered sequences in that sample was removed. OTUs remaining post-filtering were taxonomically identified using the BLASTn option within QIIME’s assign taxonomy script against the Greengenes 13.8 database. Moreover, OTUs were taxonomically assigned using RDP [31] and UCLUST [26] options, again against the Greengenes 13.8 database [27].
To determine at a gross level if there was clustering of samples according to sex and/or somatic origin, a principal co-ordinate analysis (PCoA) plot was constructed using filtered sequences negating whether or not sequences were part of the core microbiome.
Following taxonomic identification and PCoA construction, the core microbiome for each sex/somatic origin (SSO) grouping (i.e. FSH, FPH, MSH and MPH) was determined using QIIME. The ‘core’ microbiome was defined in accordance with that established by Shade et al. [32], all OTUs that occur in two or more (i.e. the majority) of the recorded time points for each of the SSO groupings. Any OTU’s occurring in only one of three time points is classed as ‘transient’ (Tr). In addition to this, the number of OTUs that were unique to an individual was determined; these were defined as all OTUs occurring solely in that individual across at least two time points irrespective of whether it was found to be a core OTU in the above SSO groupings. Upon identification of personalised OTUs, it was determined whether or not the said OTUs occurred in pubic hair, scalp hair or in both. Finally, the number of OTUs shared solely by two individuals was identified to examine whether the number of OTUs between the cohabiting couple was greater in comparison to other non-co-habiting participants.
Results and discussion
Forty-two pools of DNA extracts obtained from human scalp and pubic hairs were used to interrogate their microbial composition by next-generation sequencing. A total of 79,766 reads were generated, yielding 39,814 reads post control and abundance filtering. On average, the coverage per sample was 1,899 reads pre-filtered and 948 post-filtered. Whilst this depth of coverage is less than ideal given the advancement in NGS technology (i.e. Illumina and Ion Torrent platforms), these 454 data are still sufficient to explore the potential of hair microbial forensics for future development. Like all novel forensic techniques, metagenomic analyses of hairs will ultimately require robust evaluation and validation to ensure that these analyses are fit for purpose and able to withstand scientific scrutiny. Part of this validation should take into consideration: replication (spatial, temporal and PCR replicates); persistence of hair bacteria not only once they are transferred or deposited (during contact and stability during storage) and prevention of contamination during processing hairs in the laboratory. Budowle et al. [33] outline and discuss in detail the future validation criteria for metagenomic analyses in relation to microbial forensic applications, which they believe will require international participation. However, such an undertaking is beyond the scope of this initial evaluation into just one of many applications of forensic metagenomic investigations.
There are many ways to present metagenomic data such as generated here; the sections below explore the data using PCoA, taxonomy and OTU’s focusing on the value of the data in forensic applications. OTUs taxonomically assigned using RDP or UCLUST options revealed little to no difference in assignment to the rank of family. For this reason, all assignments refer to BLAST taxonomic assignments.
Principal coordinates plot
Of all the data generated in this study, the NGS data generated from pubic hair held the most potential for forensic applications. A general dichotomy was observed between taxa (OTUs) harboured on male and female pubic hair shafts (Figure 1).

Principal coordinate plots (PCoA). Clustering of microbial taxa from each individual at each collection time point. The lilac circle represents post-SI bacterial sequences, whilst the pale blue and yellow circles represent non-SI bacterial sequences—both circles relate solely to the co-habiting couple. Panel A represents pubic hair microbial taxa from male (orange) and female (red) participants. Panel B represents scalp hair microbial taxa from male (green) and female (blue) participants. Panel C represents microbial taxa present in male and female scalp and pubic hair samples.
In general, males were clustered close to the PC2 axis along the PC1 axis whilst females were more evenly spread along the PC1 axis and further from the PC2 axis than the males. Data relating to two individuals, who were a cohabiting couple, presented some interesting results. The red dots in the yellow ellipse at high PC1 represents the taxa present on the female partner of the couple at T0 and T2 whilst the two orange dots enclosed by small blue circles at low PC1 represent the taxa from the male partner at T0 and T2. The lilac circle encloses one red dot (taxa from the female at T5) and one orange dot (taxa from the male at T5). Microbial taxa extracted from the male and female at this time point were more similar to each other than to their other previous time points (T0, T2). Discreet enquiries revealed, unlike the preceding time points; the couple in question had engaged in sexual intercourse prior to the collection of T5 hair samples. It is noteworthy that intercourse had taken place 18 h prior to the collection of pubic hairs and both individuals had showered in the interim period. Cross-transference of bacteria during intercourse may account for the variation in taxa observed. Cross-transference, or shedding of skin micro flora, is not uncommon for individuals sharing living or communal spaces [34] or during contact sports in which Meadow et al. [35] observe ‘Our results are consistent with the hypothesis that the human skin microbiome shifts in composition during activities involving human to human contact’. The results we present here suggest that the pubic hair microbiome might be quite stable, even during cohabitation, but it might be shifted dramatically during sexual intercourse for some time. This present study is the first to suggest cross-transference of pubic/genital microbial taxa as a result of intercourse. Although further analyses need to be conducted, this initial finding bodes well for future forensic applications involving sexual crimes.
An additional advantage is that compared to other body areas such as the skin, gastrointestinal tract (GIT) and mouth, fewer bacterial species seem to comprise the vaginal microbiome [36]. The advantage of simpler communities and fewer taxa in the vaginal microbiome is one that may facilitate forensic investigations by providing results in a timely manner.
The clear microbial distinctions between pubic hairs from the sexes may largely be attributable to the prevalence of Lactobacillus spp. in the female pubic hair samples and the absence of these bacteria in the male samples (excepting the co-habiting male at T5) (Figure 1). Additionally, male pubic hair microbial taxa were clustered along axis PC2 suggesting that these taxa (OTUs) were common to the male microbiota.
In contrast, female pubic hair bacterial taxa showed elongation along axes PC1 and PC2. The elongation of data along PC1 may be attributable to females harbouring different lactobacilli species (Tables 2 and 3). However, the concomitant elongation of data along axis PC2 suggests the presence of secondary differences, differences that may be due to the presence of personalised taxa (Table 3).
The PCoA plot of male and female scalp hair microbiota over the 5-month time period did not demonstrate any significant clustering (Figure 1). This is most likely attributable to male and female scalp hairs harbouring similar bacterial taxa. However, some of the female taxa are slightly spread out along axis PC1 suggesting that there may be some variation in microbial taxa in the hairs of these individuals. The distribution and composition of the microbial communities colonising scalp and pubic hair is discussed in further detail below.
Hair microbiota
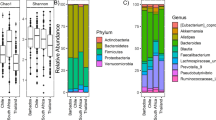
Bacteria colonising male and female scalp and pubic hair samples are classed as either ‘core’ or transient (Tr) bacteria (Figure 2, see the ‘Methods’ section). In relation to the number of OTUs extracted from scalp and pubic hair microbiomes, far less bacterial sequences were lost post control filtering for pubic microbiomes in comparison to scalp hair. Pubic hairs in general contained more OTUs than scalp hair (approximately 50 male OTUs/55 female for scalp hairs c.f. approximately 73/76 for pubic hairs). Therefore, in general, pubic hair microbiomes appear to be less influenced by environmental bacteria than scalp hairs and possible harbour more niche specific bacteria. Zhou et al. [37] support this premise by demonstrating that (in comparison to other areas of the body) vaginal microbiota consisted of less stable bacteria (i.e. more transient bacteria) and showed lower alpha diversity (i.e. low species richness), supporting the premise of pubic hair harbouring niche specific bacteria.

Microbial data extracted from scalp and pubic hairs. Diagrams illustrating core and transient (Tr) bacterial taxa on male and female scalp and pubic hair samples.
Pubic hair microbiota
Male pubic hairs could be readily distinguished from female pubic hairs on the basis of their respective microbiota. Lactobacillus was the most prevalent taxon that clearly differentiated male and female pubic hair microbiota (Figure 2). Whilst the prevalence of Lactobacillus spp. in the vagina and vaginal secretions is well established [38]-[40], this present study is the first to discuss these bacteria colonising pubic hairs, and general pubic area, in the context of probative value in forensic investigations. Fleming and Harbison [41] suggested the presence of two Lactobacillus spp. (Lactobacillus crispatus and Lactobacillus gasseri) as suitable forensic markers to identify vaginal secretions. However, microbial data garnered in this present study suggest that a NGS metagenomic approach may be preferable to those that target specific species. The variety of Lactobacillus spp. detected in pubic hairs from the female cohort consisted of 11 OTUs (taxa) in total; three Lactobacillus spp. were unique to Female 5, one Lactobacillus spp. occurred in Female 1, and four Lactobacillus spp. were uniquely between the cohabiting couple. In addition, two Lactobacillus spp. were uniquely shared between F4 and F5, and one OTU was uniquely shared between F4 and F1 (Tables 2 and 3).
Compared to male pubic hairs, female pubic hairs harboured fewer transient bacteria (Figure 2); the number of bacterial sequences comprising transient bacteria of female pubic hairs was approximately half the number of those found in male pubic hair (Table 4). This disparity may be attributable to lactobacilli conferring ‘antimicrobial protection’ to the vagina by preventing colonisation by other microorganisms [38]. Li et al. [42] also found that in comparison to other body areas, the vaginal microbiome is less transient (i.e. more stable). This stability was apparent in the differences between the number of OTUs detected in the scalp and pubic hair controls; there were significantly less OTUs present in controls from the pubic hairs in comparison to the scalp hairs. Post control filtering for FSH and MSH samples there were 33% and 43% (respectively) of sequences left. In comparison, for FPH and MPH there were 70% and 72% (respectively) sequences left, post-filtering. The disparity between the two somatic origins suggests that the bacterial taxa in scalp hair extracts had a high proportion of environmental bacteria that readily appear in controls.
Scalp hair microbiota
In contrast to the pubic hairs, scalp hair microbiota showed no correlation with the sex of the donor (Figure 2). Male and female scalp hair bacterial taxa consisted of normal human skin commensals, e.g. Anaerococcus spp., and environmentally derived taxa, e.g. Knoellia subterranea, many of which occurred in both male and female samples (Table 5). In the present study, the most significant difference observed in male and female scalp hairs was the disparate proportions of the transient bacterial taxa (Figure 2). Almost twice as many transient bacterial taxa were present in female scalp hair compared to males (Table 4). This may be due to the greater frequency of females grooming and/or washing and/or dyeing or bleaching their hair in comparison to males. Such grooming practices may prevent establishment of more stable bacterial colonies in favour of less stable (transient) bacterial colonies. Irrespective of the cause of this disparity, this observation cannot be regarded significant in relation to forensic investigations.
Costello et al. [3] identified two dominant 16S sequences from scalp swabs: Propionibacterinae in which members are predominant bacteria in hair follicles and other sebaceous sites [39] and Streptophyta (a plant phylum). In contrast, the predominant bacterial taxa from hair shafts in this study were Corynebacteriaceae and Tissierellacea fam.nov (‘new family’) (Figure 2). The difference may be attributable to either environmental differences (i.e. different study sites) or the collection technique employed by Costello et al. [3] where swabbing the top of the head might have favoured the removal of scalp/follicular bacteria (i.e. propionibacteria rather than hair shaft bacteria).
Personalised and shared bacterial taxa
Forensic investigations seek to establish ‘common origin’ or ‘source attribution’ of evidence, that is, to establish with reasonable scientific certainty that a particular individual is the source of an evidentiary sample. In relation to biological evidence, this question may be addressed through the detection of individualising biological characteristics, for example, a human DNA profile, characteristics which excludes other individuals as being the source. Ideally, these characteristics should not commonly occur within the general population or one that is solely found in males or females.
Inside the confines of the 16S V4 region, with the exception of one male (co-habiting male at T5), all individuals harboured unique taxa on their pubic hairs (Figure 3). In addition to personalised bacteria that were part of the normal skin flora, e.g. Corynebacteriaceae, pubic hairs were also colonised by environmentally derived bacterial taxa, e.g. Methylobacteriaceae (Table 3).

Personalised microbial data. Diagrammatic summary of unique bacterial taxa found in male and female scalp and pubic hair samples. Male individuals left to right: Individuals M2 (cohabiting male), M3 and M7. Female individuals left to right: Individuals F1, F4, F5 (cohabiting female) and F6.
Hairs from scalp and pubic regions, for both sexes, included shared taxa that are common inhabitants of human skin or scalp, e.g. Corynebacteria, or were environmental in origin, e.g. Rhodobacteriaceae (Tables 2 and 5). At first glance, the commonality of these bacteria may appear to be of minimal probative value; as discussed in a preceding section, personalised features should be uncommon traits or features. However, common bacteria may harbour single nucleotide polymorphisms (SNPs) within their genome, which may further discriminate between individuals.
Among all mammals, the microbiota composition is extensively conserved at the high taxonomic levels such as phylum or class. At these taxonomic levels, humans are very similar to each other (and other mammals) but variation increases progressively at the lower taxonomic levels. Personalised taxa, which allow discrimination between individuals (the goal of forensic applications), are likely to be detected at these lower taxonomic rankings. Personalised taxa may be present in high or low abundance; detection of low abundance taxa may only be detected by ultra-deep sequencing of the extracted bacterial DNA. In this regard, higher depth of coverage afforded by NGS platforms such as Ion Torrent or Illumina may be more informative than the 454 data presented here. As Ursell et al. [43] noted ‘it is important to realise that sampling depth may be critical for distinguishing taxa that are absent from those that are merely rare’. Under these circumstances, it is critical to discount bacterial taxa present in all control samples in order for the results to be not only robust but also scientifically accurate and capable of withstanding scientific and legal scrutiny.
Temporal stability data garnered in this study broadly suggest that bacteria on scalp hairs may be more prone to fluctuations in comparison to pubic hairs (in addition to being more prone to environmental contaminants). The data shows that, on average and post-filtering, approximately 17% (range 6%–25%) of pubic hair bacterial OTUs were temporally stable across all time points; whilst, on average, scalp hair harbour approximately 5% (range 0%–13%) of bacterial OTUs (Table 6). These preliminary findings suggest that pubic hair bacteria may be more temporally stable than scalp hair bacteria and therefore potentially of more probative value than scalp hair bacteria.
Although temporal stability of an individual’s bacterial taxa may appear to be an important prerequisite for metagenomics to have forensic value, the most relevant attributes will mostly likely be transference of bacteria (during contact), persistence of bacteria post transfer and storage conditions. Consider a case of unlawful sexual intercourse (of an adult female), the most relevant microbial data will be the taxa available for transfer at the time of the assault (rather than what it was weeks, months or days before or after) and the persistence of the victim’s bacteria on the offender’s genitals/pubic area (and vice versa). This, of course, is reliant upon collection of evidence from the victim and suspect(s) within several hours of the time of the assault rather than several days. Microbial data from the cohabiting couple, albeit preliminary, are encouraging, in supporting the suggestion of bacterial transfer and persistence following sexual intercourse.
Conclusions
Despite the modest sample size, we believe that the data in this qualitative assessment of metagenomic analyses of hairs are sufficient to warrant further development of this approach. For this approach to gain traction, there is a need to refine molecular targets—the broad-brush approach of the 16S V4 region looked at here is a good starting point. Additional analyses may provide further information in relation to the microbial composition of ‘core’ microbiomes and their potential value in forensic investigations. However, there is ultimately a need to develop a more focused approach that targets, for example, population level differences within Lactobacillus spp. or even more variable genomic sections of common commensals that might contain probative information at a population level.
It is suggested that microbial data gathered from hairs may provide independent data to augment other forensic results, such as mtDNA or YSTR (when DNA yields are sufficient), and possibly provide association between victims of sexual assault and offender, which is currently not possible in the absence of hairs, fibres or seminal fluid. Importantly, conducting metagenomic analyses on hairs does not preclude conducting traditional molecular analyses on the DNA extract.
Despite the complexity of microbial forensic investigations, a substrate such as hair is arguably much simpler to profile than soil a gramme of which may contain up to 50,000 different microbial species [44] or skin, which exhibits high taxonomic divergence and numbers distributed across multiple niches [45]; on the basis of our qualitative assessment, hairs harbour more modest numbers of bacterial diversity. In comparison to scalp hair, pubic hair is somewhat insulated from the environment being colonised with niche specific bacteria. With perseverance, metagenomic analyses of hairs might develop into a useful component of the forensic toolkit to augment existing forensic techniques.
Abbreviations
- BLAST:
-
basic local assignment search tool
- BLASTn:
-
Programs search nucleotide databases using a nucleotide query
- bp:
-
base pair
- emPCR:
-
emulsion-based clonal amplification
- FSH:
-
female scalp hair
- FPH:
-
female pubic hair
- GIT:
-
gastrointestinal tract
- MSH:
-
male scalp hair
- MPH:
-
male pubic hair
- MID:
-
multiplex identifier or ‘barcode’
- NGS:
-
next-generation sequencing
- qPCR:
-
quantitative polymerase chain reaction
- SI:
-
sexual intercourse
References
Parkhill J: What has high-throughput sequencing ever done for us?. Nat Rev Microbiol. 2013, 11: 664-665. 10.1038/nrmicro3112.
Structure, function and diversity of the healthy human microbiome. Nature. 2012, 486: 207-214. 10.1038/nature11234.
Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R: Bacterial community variation in human body habitats across space and time. Science. 2009, 326: 1694-1697. 10.1126/science.1177486.
Kuczynski JK, Costello EK, Nemergut ER, Zaneveld J, Labuer CL, Knights D, Koren O, Fierer N, Kelley ST, Ley RE, Gordon JI, Knight R: Direct sequencing of the human microbiome readily reveals community differences. Genome Biol 2010, 11:9.,
Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, Bonazzi V, McEwen JE, Wetterstrand KA, Deal C, Baker CC, Di Francesco V, Howcroft TK, Karp RW, Lunsford RD, Wellington CR, Belachew T, Wright M, Giblin C, David H, Mills M, Salomon R, Mullins C, Akolkar B, Begg L, Davis C, Grandison L, Humble M, Khalsa J, Little AR, et al: The NIH human microbiome project. Genome Res. 2009, 19: 2317-2323. 10.1101/gr.096651.109.
Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA, Wu D, Paulsen I, Nelson KE, Nelson W, Fouts DE, Levy S, Knap AH, Lomas MW, Nealson K, White O, Peterson J, Hoffman J, Parsons R, Baden-Tillson H, Pfannkoch C, Rogers Y-H, Smith HO: Environmental Genome Shotgun Sequencing of the Sargasso Sea. Science. 2004, 304: 66-67. 10.1126/science.1093857.
Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, Bouffard GG, Blakesley RW, Murray PR, Green ED, Turner ML, Segre JA: Topographical and temporal diversity of the human skin microbiome. Science. 2009, 324: 1190-1192. 10.1126/science.1171700.
SWGMAT: Forensic Human Hair Examination Guidelines. In Book Forensic Human Hair Examination Guidelines. 2005.
Deedrick DW, Koch SL: Microscopy of hair part 1: a practical guide and manual for human hairs. Forensic Science Communications 2004, 6.,
Houck MM: Forensic human hair examination and comparison in the 21st century. Forensic Sci Rev 2005, 17:51–66.,
Houck M, Budowle B: Correlation of microscopic and mitochondrial DNA hair comparisons. J Forensic Sci. 2002, 47: 398-475.
Linch CA, Smith SL, Prahlow JA: Evaluation of the human hair root for DNA typing subsequent to microscopic comparison. J Forensic Sci. 1998, 43: 305-314.
Opel KL, Fleishaker EL, Nicklas JA, Buel E, McCord BR: Evaluation and quantification of nuclear DNA from human telogen hairs. J Forensic Sci. 2008, 53: 853-857. 10.1111/j.1556-4029.2008.00777.x.
Butler J: Advanced Topics in Forensic DNA Methodology: San Diego, California, USA: Academic; 2011.
Nilsson M, Norlin S, Allen M: Sequencing of mtDNA in shed hairs: a retrospective analysis of material from forensic cases and a pre-screening method. Open Forensic Sci J 2012, 5:10.,
Hanson E, Haas C, Jucker R, Ballantyne J: Specific and sensitive mRNA biomarkers for the identification of skin in ‘touch DNA’ evidence. Forensic Sci Int Genet. 2012, 6: 548-558. 10.1016/j.fsigen.2012.01.004.
Mann MJ: Hair transfers in sexual assault: a six-year case study. J Forensic Sci. 1990, 35: 951-955.
Fierer N, Lauber CL, Zhou N, McDonald D, Costello EK, Knight R: Forensic identification using skin bacterial communities. Proc Natl Acad Sci U S A. 2010, 107: 6477-6481. 10.1073/pnas.1000162107.
Lauber CL, Zhou N, Gordon JI, Knight R, Fierer N: Effect of storage conditions on the assessment of bacterial community structure in soil and human-associated samples. FEMS Microbiol Lett 2010, 307:6.,
Turner S, Pryer KM, Miao VP, Palmer JD: Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J Eukaryot Microbiol. 1999, 46: 327-338. 10.1111/j.1550-7408.1999.tb04612.x.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R: Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci USA 2011, 108:7.,
Roche: Guidelines for amplicon fusion primer design for GS FLX titanium series Lib-A chemistry TCB No.013-2009. 454 Sequencing Technical Bulletin 2009, 1–3.,
Murray DC, Haile J, Dortch J, White NE, Haouchar D, Bellgard MI, Allcock RJ, Prideaux GJ, Bunce M: Scrapheap challenge: a novel bulk-bone metabarcoding method to investigate ancient DNA in faunal assemblages. Sci Rep 2013, 3:1–8.,
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A: Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012, 28: 1647-1649. 10.1093/bioinformatics/bts199.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R: QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010, 7: 335-336. 10.1038/nmeth.f.303.
Edgar RC: Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010, 26: 2460-2461. 10.1093/bioinformatics/btq461.
McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P: An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6: 610-618. 10.1038/ismej.2011.139.
Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R: PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010, 26: 266-267. 10.1093/bioinformatics/btp636.
Price MN, Dehal PS, Arkin AP: FastTree 2 - approximately maximum-likelihood trees for large alignments. PLoS One 2010, 5:1–10.,
Loman NJ, Misra RV, Dallman TJ, Constantinidou C, Gharbia SE, Wain J, Pallen MJ: Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol. 2012, 30: 434-439. 10.1038/nbt.2198.
Wang Q, Garrity GM, Tiedje JM, Cole JR: Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007, 73: 5261-5267. 10.1128/AEM.00062-07.
Shade A, Handelsman J: Beyond the Venn diagram: the hunt for a core microbiome. Environ Microbiol. 2012, 14: 4-12. 10.1111/j.1462-2920.2011.02585.x.
Budowle B, Connell N, Bielecka-Oder A, Colwell R, Corbett C, Fletcher J, Forsman M, Kadavy D, Markotic A, Morse S, Murch R, Sajantila A, Schmedes S, Ternus K, Turner S, Minot S: Validation of high throughput sequencing and microbial forensics applications. Investig Genet 2014, 5:9.,
Hospodsky D, Qian J, Nazaroff WW, Yamamoto N, Bibby K, Rismani-Yazdi H, Peccia J: Human occupancy as a source of indoor airborne bacteria. PLoS ONE 2012, 7:e34867.,
Meadow JF, Bateman AC, Herkert KM, O'Connor TK, Green JL: Significant changes in the skin microbiome mediated by the sport of roller derby. Peer J 2013, 1:e53.,
Fettweis JM, Serrano MG, Girerd PH, Jefferson KK, Buck GA: A new era of the vaginal microbiome: advances using next-generation sequencing. Chem Biodivers. 2012, 9: 965-976. 10.1002/cbdv.201100359.
Zhou Y, Gao H, Mihindukulasuriya KA, La Rosa PS, Wylie KM, Vishnivetskaya T, Podar M, Warner B, Tarr PI, Nelson DE, Fortenberry JD, Holland MJ, Burr SE, Shannon WD, Sodergren E, Weinstock GM: Biogeography of the ecosystems of the healthy human body. Genome Biol 2013, 14:R1.,
Verstraelen H, Verhelst R, Claeys G, De Backer E, Temmerman M, Vaneechoutte M: Longitudinal analysis of the vaginal microflora in pregnancy suggests that L. crispatus promotes the stability of the normal vaginal microflora and that L. gasseri and/or L. iners are more conducive to the occurrence of abnormal vaginal microflora. BMC Microbiol 2009, 9:116.,
Wilson M: Microbial Inhabitants of Humans. 2005, Cambridge University Press, Cambridge, U.K.
Witkin SS, Linhares IM, Giraldo P: Bacterial flora of the female genital tract: function and immune regulation. Best Pract Res Clin Obstet Gynaecol. 2007, 21: 347-354. 10.1016/j.bpobgyn.2006.12.004.
Fleming RI, Harbison S: The use of bacteria for the identification of vaginal secretions. Forensic Sci Int Genet. 2010, 4: 311-315. 10.1016/j.fsigen.2009.11.008.
Li K, Bihan M, Methé BA: Analyses of the stability and core taxonomic memberships of the human microbiome. PLoS ONE 2013, 8:e63139.,
Ursell LK, Metcalf JL, Parfrey LW, Knight R: Defining the human microbiome. Nutr Rev. 2012, 70 (Suppl 1): S38-S44. 10.1111/j.1753-4887.2012.00493.x.
Sensabaugh G: Microbial community profiling for the characterisation of soil evidence: forensic considerations. Criminal and Environmental Soil Forensics. Edited by: Ritz K, Dawson L, Miller D. 2009, Netherlands: Springer, New York, USA, 49-60. 10.1007/978-1-4020-9204-6_4.
Grice EA, Segre JA: The skin microbiome. Nat Rev Microbiol. 2011, 9: 244-253. 10.1038/nrmicro2537.
Alauzet C, Marchandin H, Courtin P, Mory F, Lemee L, Pons JL, Chapot-Chartier MP, Lozniewski A, Jumas-Bilak E: Multilocus analysis reveals diversity in the genus Tissierella: description of Tissierella carlieri sp. nov. in the new class Tissierellia classis nov. Syst Appl Microbiol. 2014, 37: 23-34. 10.1016/j.syapm.2013.09.007.
Ezaki T, Kawamura Y, Li N, Li ZY, Zhao L, Shu S: Proposal of the genera Anaerococcus gen. nov., Peptoniphilus gen. nov. and Gallicola gen. nov. for members of the genus Peptostreptococcus. Int J Syst Evol Microbiol. 2001, 51: 1521-1528.
Gillespie P, Hawley, PM: Principles and Practice of Clinical Bacteriology 2nd edn: John Wiley and Sons Ltd.; 2006.
Loy A, Schulz C, Lucker S, Schopfer-Wendels A, Stoecker K, Baranyi C, Lehner A, Wagner M: 16S rRNA Gene-Based Oligonucleotide Microarray for Environmental Monitoring of the Betaproteobacterial Order "Rhodocyclales". Appl Environ Microbiol. 2005, 71: 1373-1386. 10.1128/AEM.71.3.1373-1386.2005.
Fanci R, Corti, G, Bartoloni, A., Tortoli, E, Mariottini, A, Pecile, P: Unusual Methylobacterium fujisawaense Infection in a Patient with Acute Leukaemia Undergoing Hematopoietic Stem Cell Transplantation: First Case Report. Hindawi Case Reports in Medicine 2010, 2010.,
Naushad HS, Gupta RS: Molecular signatures (conserved indels) in protein sequences that are specific for the order Pasteurellales and distinguish two of its main clades. Antonie Van Leeuwenhoek. 2012, 101: 105-124. 10.1007/s10482-011-9628-4.
Valme JJMG, Leonila L, Cesareo S-J: Aurantimonas altamirensis sp. nov., a member of the order Rhizobiales isolated from Altamira Cave . International Journal of Systematic and Evolutionary Microbiology. 2006, 56: 2583-2585. 10.1099/ijs.0.64397-0.
Lo JR, Lang JM, Darling AE, Eisen JA, Coil DA: Draft genome sequence of an Actinobacterium, Brachybacterium muris strain UCD-AY4. Genome announcements 2013, 1:e0008613.,
Ivanova N, Sikorski J, Jando M, Lapidus A, Nolan M, Lucas S, Del Rio TG, Tice H, Copeland A, Cheng JF, Chen F, Bruce D, Goodwin L, Pitluck S, Mavromatis K, Ovchinnikova G, Pati A, Chen A, Palaniappan K, Land M, Hauser L, Chang YJ, Jeffries CD, Chain P, Saunders E, Han C, Detter JC, Brettin T, Rohde M, Goker M, et al: Complete genome sequence of Gordonia bronchialis type strain (3410). Stand Genomic Sci. 2010, 2: 19-28. 10.4056/sigs.611106.
Aoyama K, Kang Y, Yazawa K, Gonoi T, Kamei K, Mikami Y: Characterization of Clinical Isolates of Gordonia Species in Japanese Clinical Samples During 1998–2008. Mycopathologia. 2009, 168: 175-183. 10.1007/s11046-009-9213-9.
Allers E, Gomez-Consarnau L, Pinhassi J, Gasol JM, Simek K, Pernthaler J: Response of Alteromonadaceae and Rhodobacteriaceae to glucose and phosphorus manipulation in marine mesocosms. Environ Microbiol. 2007, 9: 2417-2429. 10.1111/j.1462-2920.2007.01360.x.
McFeters GA, Broadaway SC, Pyle BH, Egozy Y: Distribution of bacteria within operating laboratory water purification systems. Appl Environ Microbiol. 1993, 59: 1410-1415.
Srinivasan S: Managing Bacterial Regrowth and Presence in Drinking Water Distribution Systems. 2008, Ann Arbor, MI, USA, Proquest LLC
Mhedbi-Hajri N, Jacques MA, Koebnik R: Adhesion mechanisms of plantpathogenic Xanthomonadaceae. Advances in experimental medicine and biology. 2011, 715: 71-89. 10.1007/978-94-007-0940-9_5.
Garrity G, Bell J, Lilburn T: Class II. Betaproteobacteria class. nov. Bergey’s Manual® of Systematic Bacteriology. Edited by: Brenner D, Krieg N, Staley J. 2005, Springer, US, 575-922. 10.1007/0-387-29298-5_2.
Groth I, Schumann P, Schutze B, Augsten K, Stackebrandt E: Knoellia sinensis gen. nov., sp. nov. and Knoellia subterranea sp. nov., two novel actinobacteria isolated from a cave. Int J Syst Evol Microbiol. 2002, 52: 77-84.
Urakami T, Oyanagi H, Araki H, Suzuki K-I, Komagata K: Recharacterization and Emended Description of the Genus Mycoplana and Description of Two New Species, Mycoplana ramosa and Mycoplana segnis. International journal of systematic bacteriology. 1990, 40: 434-442. 10.1099/00207713-40-4-434.
Acknowledgements
The authors would like to thank the volunteers for their participation, diligence and patience. We thank the Murdoch University Centre for comparative genomics (M. Bellgard) and iVEC computing resource. MB was supported in this research by an ARC future fellowship FT0991741.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SRT and MB conceived and designed the experiments. SRT and JA performed the experiments. SRT, DM, KPK, and MB performed the data analysis and wrote the paper. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Tridico, S.R., Murray, D.C., Addison, J. et al. Metagenomic analyses of bacteria on human hairs: a qualitative assessment for applications in forensic science. Investig Genet 5, 16 (2014). https://doi.org/10.1186/s13323-014-0016-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13323-014-0016-5