
Abstract
Mood disorders are devastating, often chronic illnesses characterized by low mood, poor affect, and anhedonia. Notably, mood disorders are approximately twice as prevalent in women compared to men. If sex differences in mood are due to underlying biological sex differences, a better understanding of the biology is warranted to develop better treatment or even prevention of these debilitating disorders. In this review, our goals are to: 1) summarize the literature related to mood disorders with respect to sex differences in prevalence, 2) introduce the corticolimbic brain network of mood regulation, 3) discuss strategies and challenges of modeling mood disorders in mice, 4) discuss mechanisms underlying sex differences and how these can be tested in mice, and 5) discuss how our group and others have used a translational approach to investigate mechanisms underlying sex differences in mood disorders in humans and mice.
Similar content being viewed by others

Review
Sex differenced in major depression
Major depressive disorder (MDD) is a severe mental illness and the leading cause of disability and of years of productivity lost worldwide [1]. In addition to the psychological stress on patients and families, MDD contributes to the development and progression of systemic and organ diseases [2]-[5]. For instance, MDD increases the risk for coronary heart disease incidence by approximately 1.7 times compared to non-depressed subjects [6], and MDD patients have a 37% increased risk for developing type 2 diabetes [7]. Moreover, patients with mood disorders (MDD or bipolar disorder) make up approximately 60% of completed suicides [8]. MDD is defined as a syndrome that includes prominent emotion dysregulation, low mood, poor affect, and/or anhedonia; these core MDD symptoms are accompanied by cognitive symptoms (attention, concentration), physiological symptoms (weight, locomotor, and sleep pattern changes) [9], and frequent co-morbid high anxiety symptoms [9],[10].
Notably, women are twice as likely to be diagnosed with MDD compared to men [11],[12]. When men and women that have been diagnosed with MDD are compared, women tend to have more symptoms and higher symptom severity, and women report more subjective distress [13]-[15]. Additionally, anxiety symptoms are almost always co-morbid with MDD in women, making the two difficult to separate. In fact, women are more likely than men to have a co-morbid anxiety disorder with MDD (e.g., [16]), and men more likely to have a co-morbid substance abuse disorder (reviewed in [17]), possibly suggesting different coping strategies in males and females. A frequent and important question is whether the sex difference in MDD incidence is an artifact of women being more likely to seek treatment. However, this sex difference in MDD incidence is consistently found across cultures and in community-based epidemiological studies, in which the factor of seeking treatment is removed (e.g., [13],[18]), suggesting that there are biological differences that place women at increased risk for MDD. Also arguing against the potential artifact of women being more likely to seek treatment, Bogner and Gallo found no sex difference in self-report of depressive symptoms in a community-based epidemiological study [19].
Some studies suggest that women respond differently than men to antidepressant treatment. For instance, a study by Kornstein et al. [11] found that women responded more favorably (i.e., reduced symptoms, fewer adverse effects) to selective serotonin reuptake inhibitors (SSRIs) than men; conversely, men responded more favorably than women to tricyclic antidepressants [11],[20]. However, other studies have reported no sex differences in response to SSRIs or tricyclics (e.g., [21]), but a statistically superior response to monoamine oxidase inhibitors (MAOIs) in women compared to men [21]. Entsuah et al. [22] found no sex difference in response to SSRIs or to venlafaxine, a serotonin norepinephrine reuptake inhibitor. Taken together, this suggests variable or no sex differences in antidepressant response, specifically compared to the robust and replicated findings of sex differences in symptom dimensions.
It has been proposed that the increased prevalence of MDD in women may be due to how women perceive stress [23]. In other words, women may have the “trait” of having more subjective distress in stressful situations compared to men. Indeed, even when considering men and women without an MDD diagnosis, there are sex differences in response to stressful situations. For instance, even when men and women have equivalent physiological responses to the same stressful situation (no differences in heart rate or plasma cortisol), women self-report higher irritability and fear as well as decreased happiness compared to men [23].
Depression-related sex differences in a corticolimbic network of mood regulation
Even though the neurobiological mechanism(s) underlying MDD remain poorly characterized, evidence from both neuroimaging and postmortem neuroanatomical and molecular studies suggest a dysfunction in the emotion regulation centers of the brain underlying low affect, a symptom dimension common to both MDD and anxiety disorders [24]-[27]. This corticolimbic network includes the prefrontal and anterior cingulate cortices, the hippocampus, the anterior thalamic nuclei, and the amygdala [24],[28]. The subgenual anterior cingulate cortex (sgACC) consistently shows elevated metabolic activity with the induction of depressive states [29]-[31], which returns to normal following antidepressant treatment [30] or deep brain stimulation [32]. Interestingly, neuroimaging studies show that features of sgACC dysfunction in MDD are sexually dimorphic, with women exhibiting higher levels of reactivity compared to males [33]-[35]. The amygdala processes emotionally salient stimuli and, in concert with cortical and subcortical interconnections, initiates a behavioral response [27]. Neuroimaging studies show that MDD patients exhibit abnormal processing of emotional stimuli, with sustained amygdala reactivity [36],[37] (although [38],[39]). Similarly, amygdala hyperactivity is reported in patients with various anxiety disorders, including post-traumatic stress, generalized anxiety, and social anxiety disorders [40].
How do we model mood disorders in mice?
Validity of animal models
When assessing any animal model of a psychiatric disorder, several criteria need to be considered. The animal model should have construct validity, that is, it should follow a similar etiology as the human disorder. The model should have face validity, with anatomical, behavioral, or molecular features of the disorder being replicated. Predictive validity should also be considered, as pharmacological treatment in the animal model should recapitulate the effects of treatment in humans. Importantly, both the effect and time-course of efficacious treatments in humans need to be taken into account when assessing predictive validity of an animal model.
Trait versus state
When researchers investigate anxiety-/depressive-like behavior in mice, they often do so under baseline (i.e., “trait”) conditions. These traits represent properties of the biological and behavioral system that may play a role in susceptibility to develop a psychiatric disorder. On the other hand, MDD represents a temporary mood “state”; in other words, MDD can be considered a transient pathological state that is brought on by certain factors, i.e., depressive episodes. Studies under baseline conditions are relevant, as they can provide insight into potential predisposition for developing an MDD state. We argue, however, that many studies only examine trait conditions. We feel that this is especially true in mouse studies investigating the origin of sex differences in anxiety-/depressive-like behaviors. There are, however, models that researchers can use to examine these anxiety-/depressive-like behaviors under pathological state conditions that are homologous to depressive episodes.
One model that is used to study mice in an elevated mood-related state is unpredictable chronic mild stress (UCMS). UCMS was originally developed in rats, and our lab and others have recently used UCMS in mice to model human MDD episodes. UCMS replicates the role of stress in eliciting MDD, with rodents developing a depressive-like syndrome after several weeks of random exposure to mild social and environmental stressors. Specifically, these mice have heightened fearfulness/anxiety-like behavior [41], anhedonia-like behavior, as assessed by decreased consumption of palatable food and drink [41],[42] and decreased sensitivity to rewards [43], and physiological symptoms (decreased weight gain and grooming behavior (e.g., [44]). Additionally, there is dysregulation of the hypothalamic pituitary adrenal (HPA) axis and elevated basal plasma corticosterone [45], as reported in some MDD patients (e.g., [46]-[49]). The UCMS syndrome respects the time frame of onset and efficacy of antidepressant treatment [42],[50],[51]. Interestingly, not all mice exhibit a depressive-like syndrome following UCMS exposure, making it more realistic, as differences in response to stress exposure are also observed in humans (e.g., [52]), and making it a potential model to study both vulnerability and resiliency to develop a depressive-like episode. One significant drawback of UCMS is that it is not a simple procedure to perform: it is labor intensive and lasts for 4–9 weeks. Additionally, UCMS is not as highly reproducible as some other mouse models of MDD (e.g., genetic models), and this may be due to a number of factors, including among others varying stress procedures, duration of UCMS, strain of mice used, and normal heterogeneity in stress response.
Another mouse model that elicits an elevated mood state is chronic social defeat stress. With this paradigm, male rodents are subjected to repeated bouts of social subordination [53]. There are several benefits of this paradigm: 1) it has construct validity (chronic stress elicits the behavioral deficits), 2) it has predictive validity (chronic antidepressant treatment reverses behavioral deficits), 3) it affects multiple systems as MDD does (e.g., dopaminergic reward circuits and hippocampal neurotrophin), and 4) it is useful to study mechanisms underlying resilience (e.g., [54]). However, there is a challenge with the chronic social defeat paradigm when one is interested in sex differences in MDD, since this paradigm seems to only be effective in male, but not female C57BL/6 mice. Researchers have got around this limitation by using different species of mice or by using rats (e.g., [55]-[57]). The social defeat paradigm has been used successfully in the monogamous California mouse (Peromyscus californicus), in which both males and females aggressively defend territories [58]. Interestingly, Trainor and colleagues [59] reported no effect of adult hormone manipulation in the paradigm, but an effect of corncob bedding (which has estrogenic properties) during development, together suggesting developmental hormonal programming.
Learned helplessness is another model used in the rodent literature to induce a depressive-like state [60],[61]. In this model, the rodent is exposed to a noxious stimulus (often a shock) that it either can or cannot escape. When later tested under conditions in which escape is possible, the rodent that previously was exposed to the inescapable shock often does not learn to escape. Importantly, the learned helplessness model has: 1) construct validity (uncontrollable stressful events precipitate the deficit), 2) predictive validity (improved response after antidepressant treatment), and 3) face validity (equated with the helplessness experienced by humans with MDD). A very interesting aspect of the learned helplessness model is that it seems to be ineffective in eliciting a depressive-like state in female rats [62] and in female C57BL/6 mice [63]. This sex difference may be strain/species specific, as both male and female 129SvEv mice develop learned helplessness [63]. Notably, the sex difference in learned helplessness in rats was not reversed after removal of adult hormones by gonadectomy [62], suggesting either developmental hormonal or sex chromosome complement effects.
Modeling sex differences in mice
When a sex difference is observed, there are several steps that can be taken to determine the cause(s) of the sex difference. There are several comprehensive reviews on this topic (e.g., [64]-[66]); we summarize the general strategy here. The first and easiest step is to test whether the sex difference disappears after normalizing, or “clamping”, circulating gonadal hormones between males and females. This can be accomplished by simply gonadectomizing (GDX) adult males and females. If the sex difference is no longer present after GDX, we know that the sex difference was caused by the differences in circulating hormones between males and females. Sex differences that disappear when circulating hormones are made equivalent between males and females are said to be due to “activational” effects of gonadal hormones.
If the observed sex difference persists even when males and females have the same circulating hormone exposure, the next logical step is to test whether the sex difference is influenced by developmental hormone exposure (i.e., “organizational” effects of hormones). Here, exposure to gonadal hormones during critical developmental periods causes permanent effects on the body, and these sex differences persist when adult hormones are made equivalent. The concept of the critical developmental window is actually quite tricky, as this window is not necessarily the same for every trait examined, and the window can extend from the prenatal into the postnatal period. Additionally, with respect to reproductive behavior, testosterone exposure during development performs both a masculinizing (organization of the neural control mechanisms for adult male sex behavior) and a defeminizing function (loss of ability to respond to the activational effects of ovarian hormones to induce female sex behavior); notably, testosterone may perform these organizational effects during different critical windows (see reviews [67],[68]). Testing for organizational effects of hormones can be accomplished in a few different ways. One commonly used method is to treat females with a dose of testosterone similar to what males are normally exposed to during a critical developmental period; this critical period is typically thought to be right around the time of birth in rodents (but prenatally in some species; reviewed in [69]). If the females treated developmentally with testosterone are not significantly different from normal males, then the sex difference was due to organizational effects of hormones. Another method for testing for organizational effects of hormones is to remove the developmental testosterone exposure in males by GDX during the critical developmental period and determining whether these males are significantly different from normal females. However, this developmental GDX method is technically more challenging, as the procedure would have to take place prenatally, and it is difficult to know whether hormones were completely removed during the critical developmental window. There are several important questions to consider with studies aimed at manipulating hormone exposure during critical developmental windows: 1) Does incomplete masculinization or defeminization mean that the sex difference examined is not programmed by developmental hormone exposure or was the critical developmental window partially missed?, and 2) Is a single dose of testosterone enough for complete masculinization/defeminization or is prolonged exposure necessary? Notably, recent studies have identified puberty as an additional critical period for organizational effects of gonadal hormones (reviewed in [70]).
If the observed sex difference persists even after manipulating developmental hormone exposure, the next step is to test for potential effects of the sex chromosome complement. Genetic males have only one X chromosome and one Y chromosome, while genetic females have two X chromosomes. Thus, genes on the Y chromosome or gene dosage of the X chromosome could play a role in sexual dimorphism (reviewed in [71]). Even though researchers knew as early as the 1950s that the presence of the Y chromosome caused the undifferentiated gonads to develop into testes [72], work in the 1990s zeroed in on the SRY gene (Sry in mice) as being the testis-determining gene [73],[74]. The testes in turn produce androgens to drive differentiation of the male internal and external genitalia. In the absence of the Y chromosome, and therefore lack of SRY/Sry gene product, the undifferentiated gonads develop into ovaries [72]. Since the testis-determining gene (Sry) is found on the Y chromosome, it is impossible to separate the potential role of sex chromosome complement from gonadal (and therefore, hormonal sex) in traditional wild-type mice, regardless of hormone manipulation. Thus, genetic manipulation has been used to engineer the four core genotypes (FCG) mice, in which Sry has been placed on an autosome after spontaneous deletion from the Y chromosome. Thus, genetic and gonadal sex are dissociated in the FCG mice, and the contribution of sex chromosome complement can be investigated independently (reviewed in [75]). Using the FCG mice, investigators can independently assess the contribution of sex chromosome complement, developmental hormone exposure, and adult circulating hormones to various observed sex differences. Importantly, to be able to probe for potential developmental hormone effects, all mice must be GDX several weeks prior to examination, such that any differences observed due to gonads are considered to be permanent changes due to hormone exposure during a critical period of development. A key strength of the FCG model is its ability to identify potential organizational hormone effects during the perinatal life and during puberty. Another mouse model that is useful for studying the contribution of sex chromosome complement to sex differences is the steroidogenic factor 1 (SF-1) KO mouse. SF-1 (encoded by the Nr5a1 gene) is a transcription factor involved in the reproductive system and is normally expressed in the gonads, adrenal cortex, pituitary gland, and ventromedial nucleus of the hypothalamus [76],[77]. SF-1 KO mice lack gonads and therefore also lack endogenous gonadal hormones during development and in adulthood; thus, SF-1 KO mice are useful to examine the effects of sex chromosome without the potential confounding effects of endogenous gonadal hormones [78]-[82].
Once sex chromosome complement has been identified as a contributing factor to the sex difference of interest, it is often important to determine whether the dosage of X chromosomes or the presence of the Y chromosome underlies the sex difference. To this end, researchers use the Y* mice, which have varying numbers of X and Y chromosomes, and Y chromosome consomic strains, in which the strains are genetically identical except for the Y chromosome (reviewed in [83]).
Of mice and men: how do we investigate sex differences in mood disorders?
Humans
Our lab and others have reported numerous differences in the postmortem brains of MDD patients compared to healthy controls. The goal of these studies is to identify genes and proteins that are altered in the brains of MDD patients in order to identify factors that may cause MDD. Recent postmortem molecular studies [84]-[86] support the hypothesis of a deficit in inhibitory neurotransmission in MDD. Specifically, reduced expression of somatostatin (SST), a marker for inhibitory gamma-aminobutyric acid (GABA) neurons targeting pyramidal cell dendrites was observed in several brain regions in the corticolimbic network of mood regulation [sgACC [87], amygdala [86], and dorsolateral prefrontal cortex (DLPFC) [88]]. In concert, these findings suggest a GABA/SST-related cellular phenotype of reduced dendritic inhibition in depression. Using meta-analysis and meta-regression in eight human postmortem microarray studies in DLPFC, sgACC, and amygdala, we confirmed that SST is significantly decreased in subjects with MDD compared to matched controls and importantly, showed that the SST reduction in female MDD is significantly more robust than results in male MDD [89], together demonstrating a sexual dimorphism in reduced SST in MDD.
Another method that we have used in the human postmortem brain is gene co-expression analysis. Co-expression is defined as correlated gene expression across samples and has been shown to reflect shared gene function, including common regulation (e.g., hormones, transcription). The goal of these studies is to assess the broader biological context associated with our genes of interest. Using SST as our “seed” gene of interest, we identified GABA receptor signaling and mitochondrial dysfunction as the top canonical pathways represented by genes co-expressed with SST. Notably, this top 200 SST-co-regulated gene selection included GABA synthesizing enzymes glutamate decarboxylase 1 (GAD1; also known as GAD67) and GAD2 (also known as GAD65), hence confirming the functional relevance of an SST/GABA-related biological module [89]. Combined with our findings of a more robust reduction in SST in women with MDD, these gene co-expression studies suggest that more robust GABA-related deficits may characterize female MDD.
A major roadblock that we encounter when using human postmortem brains is that we often do not have blood samples from the same subjects, making circulating gonadal hormone analysis impossible. To partially circumvent this limitation, we have combined gene expression analysis and single nucleotide polymorphism (SNP) genotyping in the same subjects [i.e., expression quantitative trail loci (eQTL) study]. In these eQTL studies, we searched for SNPs (i.e., genetic polymorphisms) that are associated with either increased or decreased expression of our genes of interest. For instance, we performed a targeted eQTL study to test the hypothesis of X chromosome genetic contribution to SST, GAD1, and GAD2 gene expression. Even though the SST, GAD1, and GAD2 genes are not located on the X chromosome, we found several X chromosome SNPs associated with expression of these three genes; these results suggest the possibility of trans-regulation of SST, GAD1, and GAD2 by X chromosome-encoded factors [89]. Together, these correlative findings provide support for a contribution of genetic sex to sexual dimorphism in affect dysregulation in human subjects, potentially mediated by X chromosome trans-regulation of key GABA-related genes.
Mice
Although studies in the human postmortem brain have been highly informative in uncovering potential leads for the molecular mechanism underlying female vulnerability to MDD, complementary studies in mice are necessary to test for mechanisms underlying observed human sex differences. Importantly, we recently showed that the UCMS paradigm recapitulates the female vulnerability to MDD. Although both male and female UCMS-exposed mice developed elevated anxiety- and depressive-like behaviors compared to non-stressed controls, the chronically stressed females (which were freely cycling) exhibited a more robust elevation in behavioral emotionality compared to chronically stressed males [90], thus providing a needed assay to investigate the sexual dimorphic bases of human MDD. Here, “behavioral emotionality” or “emotionality” is defined as combined and measurable anxiety- and depressive-like behaviors in mice. Importantly, we did not find an interaction between UCMS exposure and sex, suggesting similar underlying mechanisms in males and females, but with additional factors at play in one or both sexes.
In a related experiment, we aimed to determine the potential contribution of developmental and adult hormone exposure to our observed sex difference in response to chronic stress exposure. To examine the potential developmental organizational role of hormones in establishing adult sex differences in emotionality, we tested the impact of neonatal testosterone exposure (a validated approach to developmentally “masculinize” the brain) [91]-[94] on adult emotionality in mice. We also examined potential activational hormone effects by comparing mice from each neonatal group that were GDX in adulthood and implanted with estradiol capsules or given sham surgery and blank implants. Results indicated that neonatal testosterone exposure partially masculinized UCMS-induced high emotionality of female mice; the females treated neonatally with testosterone displayed emotionality measures intermediate between normal males and females. Overall, we did not observe consistent activational effects of estradiol, although these studies were not designed to maximize these contrasts. Notably, other studies have reported effects of adult circulating hormones on emotionality (e.g., [95],[96]). Indeed, Laplant and colleagues [97] reported that GDX of females prevents the pro-depressive-like effects of chronic stress. Our results in females treated neonatally with testosterone suggested that another factor, potentially sex chromosome complement, could influence the observed sex difference in emotionality. Although female mice in this study were treated neonatally with testosterone, mirroring the developmental testosterone exposure experienced by males, they were still genetically female for their entire lives; this suggests that genetic sex, regardless of developmental or adult hormone exposure, represents an additional factor contributing to adult emotionality. It is important to note that these females treated neonatally with testosterone did not have adult testosterone levels equivalent to a normal male. Thus, a “cleaner” model (e.g., FCG mice) is necessary to disentangle the potential effects of gonadal and genetic sex on emotionality.
Following up on the hint of a role for genetic sex, we used the FCG mice in a next set of experiments as a tool to separate the potential contributions of developmental hormone exposure, adult hormone exposure, and sex chromosome complement to adult emotionality. FCG mice were GDX as adults to remove endogenous gonadal hormones and implanted with testosterone-filled or blank capsules to also investigate the activational effects of testosterone. We then assessed anxiety-like behavior (using elevated plus maze and open field) under baseline (no stress) conditions and after exposure to UCMS. Under baseline conditions, the sex-related factor influencing emotionality was sex chromosome complement; however, the effect was in the opposite direction to what we had predicted based on the female vulnerability to mood disorders. Specifically, XY mice, regardless of gonadal sex or adult circulating testosterone treatment, exhibited increased anxiety-like behavior relative to XX mice. This sex chromosome effect was amplified after UCMS exposure. Additionally, we saw a potent effect of circulating testosterone to decrease anxiety-like behavior in UCMS-exposed mice, consistent with prior evidence in the literature [98]. Developmental hormone exposure had inconsistent effects on anxiety-like behaviors under both no stress and UCMS conditions. Although we reported a sex chromosome effect for the first time for anxiety-like behavior, a conventional interpretation in the sex difference field is that XY sex chromosome complement may exert a compensatory effect to reduce differences otherwise induced by circulating testosterone or vice versa [99]; indeed, FCG studies have reported similar opposing actions of XY and circulating testosterone [100],[101]. Since “intact” male mice exhibit lower emotionality than females [90] and since we observed a more robust effect of circulating testosterone on lowering anxiety-like behaviors compared to the anxiogenic effect of XY genetic sex, circulating testosterone seems to “win out” in a normal male; the end result being lower anxiety-like behavior in males [89]. The robust behavioral findings and opposing effects of XY genetic sex and circulating testosterone demonstrate that both factors critically contribute to a dynamic equilibrium regulating adult anxiety-like behaviors.
To begin to search for the molecular underpinnings of the opposing effects of male sex chromosome complement and circulating testosterone on anxiety-like behavior, we examined expression of several mood-related genes in the frontal cortex of FCG mice. Specifically, we began by examining expression of several genes related to GABA, serotonin, and dopamine signaling, as candidate systems implicated in mood disorders. Several studies suggest impaired excitation/inhibition balance in mood disorders, potentially mediated by decreased GABA inhibition [84],[86]-[88],[102]-[106]. Additionally, results also suggest problems with slow-acting serotonin and dopamine neuromodulatory systems in mood disorders [107]-[115]. Interestingly, results showed that the sex-related factor that had the strongest effect on expression of these mood-related genes was sex chromosome complement. Overall, mice with XY sex chromosome complement tended to have lower expression of these GABA-, serotonin-, and dopamine-related genes compared to XX mice. These gene expression findings correlated nicely with our finding that XY mice also had elevated anxiety-like behavior. Developmental hormone exposure resulted in varied effects: mice with male hormone exposure during development had higher expression of GABA-related genes but lower expression of serotonin- and dopamine-related genes. Adult testosterone exposure exhibited inconsistent effects [116]. Together, these studies provided some molecular support to the behavioral studies investigating the contribution of XY sex chromosome complement to adult behavioral emotionality.
Conclusions
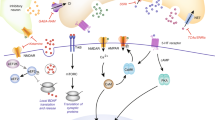
There is clear evidence that women are more vulnerable to develop mood disorders compared to men. This sex difference seems to have a biological basis, as we have found sex differences in expression of mood-related genes in the brains of depressed subjects. Our work thus far suggests that a dynamic equilibrium exists between the effects of male sex chromosome complement to increase anxiety, which is opposed by the antianxiety effects of male circulating testosterone exposure. Figure 1 provides a schematic summarizing our interpretation of the findings described in the previous sections. Specifically, our work in humans and in mice shows that sex chromosome complement influences expression of SST and other GABA-related genes [89],[116]. Our mouse studies also show that while testosterone has a potent effect of decreasing anxiety-like behavior, it does not seem to be doing so via effects on GABA-, serotonin-, or dopamine-related gene expression [89],[116]. We hypothesize that testosterone acts to oppose the pro-anxiety effects of male sex chromosome complement by affecting the function of SST cells and/or the function of the local cortical microcircuitry (Figure 1). Finally, we believe that preliminary studies using appropriate mouse models, with consideration of trait and state, as well as the multiple dimensions of mood-related behaviors, can provide a framework to systematically dissect the biological underpinnings of sex differences in mood in humans.

A novel model depicting a dynamic balance between sex chromosome complement and circulating testosterone on anxiety (black dotted line). A conventional interpretation in the sex difference field is that XY sex chromosome complement may exert a compensatory effect to reduce differences otherwise induced by circulating testosterone or vice versa. Data outlined in this review suggests that this is also the case for the regulation of mood and anxiety-like behaviors. Specifically, while male sex chromosome complement causes an increase in anxiety-like behavior, this effect is opposed by testosterone’s antianxiety effect. While sex chromosome complement appears to be acting via control of gene expression, we hypothesize that testosterone affects the activity of certain inhibitory GABA cell types (e.g., SST cells).
Abbreviations
- DLPFC:
-
dorsolateral prefrontal cortex
- eQTL:
-
expression quantitative trait loci
- FCG:
-
four core genotypes
- GABA:
-
gamma-aminobutyric acid
- GAD1:
-
glutamate decarboxylase 1
- GAD2:
-
glutamate decarboxylase 2
- GAD65:
-
glutamate decarboxylase 65
- GAD67:
-
glutamate decarboxylase 67
- GDX:
-
gonadectomize
- HPA:
-
hypothalamic pituitary adrenal
- MDD:
-
major depressive disorder
- SF-1:
-
steroidogenic factor 1
- sgACC:
-
subgenual anterior cingulate cortex
- SNP:
-
single nucleotide polymorphism
- SSRI:
-
selective serotonin reuptake inhibitor
- SST:
-
somatostatin
- UCMS:
-
unpredictable chronic mild stress
References
WHO:World Health Organization - The Global Burden of Disease - 2004 Update. WHO Library, Geneva, Switzerland; 2004.
Murray CJ, Lopez AD: Global mortality, disability, and the contribution of risk factors: global burden of disease study. Lancet 1997, 349: 1436–1442.
Musselman DL, Evans DL, Nemeroff CB: The relationship of depression to cardiovascular disease: epidemiology, biology, and treatment. Arch Gen Psychiatry 1998, 55: 580–592.
Michelson D, Stratakis C, Hill L, Reynolds J, Galliven E, Chrousos G, Gold P: Bone mineral density in women with depression. N Engl J Med 1996, 335: 1176–1181.
Schulz R, Beach SR, Ives DG, Martire LM, Ariyo AA, Kop WJ: Association between depression and mortality in older adults: the cardiovascular health study. Arch Intern Med 2000, 160: 1761–1768.
Ferketich AK, Schwartzbaum JA, Frid DJ, Moeschberger ML: Depression as an antecedent to heart disease among women and men in the NHANES I study. National health and nutrition examination survey. Arch Intern Med 2000, 160: 1261–1268.
Knol MJ, Twisk JW, Beekman AT, Heine RJ, Snoek FJ, Pouwer F: Depression as a risk factor for the onset of type 2 diabetes mellitus. A meta-analysis. Diabetologia 2006, 49: 837–845.
Mann JJ: Neurobiology of suicidal behaviour. Nat Rev Neurosci 2003, 4: 819–828.
Belmaker RH, Agam G: Major depressive disorder. N Engl J Med 2008, 358: 55–68.
Kendler KS, Prescott CA, Myers J, Neale MC: The structure of genetic and environmental risk factors for common psychiatric and substance use disorders in men and women. Arch Gen Psychiatry 2003, 60: 929–937.
Kornstein SG, Schatzberg AF, Thase ME, Yonkers KA, McCullough JP, Keitner GI, Gelenberg AJ, Ryan CE, Hess AL, Harrison W, Davis SM, Keller MB: Gender differences in chronic major and double depression. J Affect Disord 2000, 60: 1–11.
Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE: Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the national comorbidity survey replication. Arch Gen Psychiatry 2005, 62: 593–602.
Angst J, Dobler-Mikola A: Do the diagnostic criteria determine the sex ratio in depression? J Affect Disord 1984, 7: 189–198.
Frank E, Carpenter LL, Kupfer DJ: Sex differences in recurrent depression: are there any that are significant? Am J Psychiatry 1988, 145: 41–45.
Young MA, Fogg LF, Scheftner WA, Keller MB, Fawcett JA: Sex differences in the lifetime prevalence of depression: does varying the diagnostic criteria reduce the female/male ratio? J Affect Disord 1990, 18: 187–192.
Silverstein B: Gender difference in the prevalence of clinical depression: the role played by depression associated with somatic symptoms. Am J Psychiatry 1999, 156: 480–482.
Najt P, Fusar-Poli P, Brambilla P: Co-occurring mental and substance abuse disorders: a review on the potential predictors and clinical outcomes. Psychiatry Res 2011, 186: 159–164.
Weissman MM, Klerman GL: Sex-differences and epidemiology of depression. Arch Gen Psychiatry 1977, 34: 98–111.
Bogner HR, Gallo JJ: Are higher rates of depression in women accounted for by differential symptom reporting? Soc Psychiatry Psychiatr Epidemiol 2004, 39: 126–132.
Hamilton JA, Grant M, Jensvold MF: Sex and treatment of depression: When does it matter? In Psychopharmacology and Women: Sex, Gender, and Hormones. Edited by: Jensvold MF, Halbreich U, Hamilton JA. American Psychiatric Press, Washington, DC; 1996:241–257.
Quitkin FM, Stewart JW, McGrath PJ, Taylor BP, Tisminetzky MS, Petkova E, Chen Y, Ma G, Klein DF: Are there differences between women’s and men’s antidepressant responses? Am J Psychiatry 2002, 159: 1848–1854.
Entsuah AR, Huang H, Thase ME: Response and remission rates in different subpopulations with major depressive disorder administered venlafaxine, selective serotonin reuptake inhibitors, or placebo. J Clin Psychiatry 2001, 62: 869–877.
Kelly MM, Tyrka AR, Anderson GM, Price LH, Carpenter LL: Sex differences in emotional and physiological responses to the trier social stress test. J Behav Ther Exp Psychiatry 2008, 39: 87–98.
Seminowicz DA, Mayberg HS, McIntosh AR, Goldapple K, Kennedy S, Segal Z, Rafi-Tari S: Limbic-frontal circuitry in major depression: a path modeling metanalysis. Neuroimage 2004, 22: 409–418.
Mayberg HS: Limbic-cortical dysregulation: a proposed model of depression. J Neuropsychiatry Clin Neurosci 1997, 9: 471–481.
Gaiteri C, Guilloux JP, Lewis DA, Sibille E: Altered gene synchrony suggests a combined hormone-mediated dysregulated state in major depression. PLoS One 2010, 5: e9970.
Martin EI, Ressler KJ, Binder E, Nemeroff CB: The neurobiology of anxiety disorders: brain imaging, genetics, and psychoneuroendocrinology. Clin Lab Med 2010, 30: 865–891.
Pezawas L, Meyer-Lindenberg A, Drabant EM, Verchinski BA, Munoz KE, Kolachana BS, Egan MF, Mattay VS, Hariri AR, Weinberger DR: 5-HTTLPR polymorphism impacts human cingulate-amygdala interactions: a genetic susceptibility mechanism for depression. Nat Neurosci 2005, 8: 828–834.
Drevets WC, Ongur D, Price JL: Reduced glucose metabolism in the subgenual prefrontal cortex in unipolar depression. Mol Psychiatry 1998, 3: 190–191.
Mayberg HS, Brannan SK, Tekell JL, Silva JA, Mahurin RK, McGinnis S, Jerabek PA: Regional metabolic effects of fluoxetine in major depression: serial changes and relationship to clinical response. Biol Psychiatry 2000, 48: 830–843.
Siegle GJ, Thompson W, Carter CS, Steinhauer SR, Thase ME: Increased amygdala and decreased dorsolateral prefrontal BOLD responses in unipolar depression: related and independent features. Biol Psychiatry 2007, 61: 198–209.
Mayberg HS, Lozano AM, Voon V, McNeely HE, Seminowicz D, Hamani C, Schwalb JM, Kennedy SH: Deep brain stimulation for treatment-resistant depression. Neuron 2005, 45: 651–660.
Craig AD: How do you feel–now? The anterior insula and human awareness. Nat Rev Neurosci 2009, 10: 59–70.
Drevets WC, Savitz J, Trimble M: The subgenual anterior cingulate cortex in mood disorders. CNS Spectr 2008, 13: 663–681.
Vogt BA: Pain and emotion interactions in subregions of the cingulate gyrus. Nat Rev Neurosci 2005, 6: 533–544.
Siegle GJ, Steinhauer SR, Thase ME, Stenger VA, Carter CS: Can’t shake that feeling: event-related fMRI assessment of sustained amygdala activity in response to emotional information in depressed individuals. Biol Psychiatry 2002, 51: 693–707.
Sheline YI, Barch DM, Donnelly JM, Ollinger JM, Snyder AZ, Mintun MA: Increased amygdala response to masked emotional faces in depressed subjects resolves with antidepressant treatment: an fMRI study. Biol Psychiatry 2001, 50: 651–658.
Townsend JD, Eberhart NK, Bookheimer SY, Eisenberger NI, Foland-Ross LC, Cook IA, Sugar CA, Altshuler LL: fMRI activation in the amygdala and the orbitofrontal cortex in unmedicated subjects with major depressive disorder. Psychiatry Res 2010, 183: 209–217.
Monk CS, Klein RG, Telzer EH, Schroth EA, Mannuzza S, Moulton JL 3rd, Guardino M, Masten CL, McClure-Tone EB, Fromm S, Blair RJ, Pine DS, Ernst M: Amygdala and nucleus accumbens activation to emotional facial expressions in children and adolescents at risk for major depression. Am J Psychiatry 2008, 165: 90–98.
Engel K, Bandelow B, Gruber O, Wedekind D: Neuroimaging in anxiety disorders. J Neural Transm 2009, 116: 703–716.
Pothion S, Bizot JC, Trovero F, Belzung C: Strain differences in sucrose preference and in the consequences of unpredictable chronic mild stress. Behav Brain Res 2004, 155: 135–146.
Muscat R, Willner P: Suppression of sucrose drinking by chronic mild unpredictable stress: a methodological analysis. Neurosci Biobehav Rev 1992, 16: 507–517.
Papp M, Willner P, Muscat R: An animal model of anhedonia: attenuation of sucrose consumption and place preference conditioning by chronic unpredictable mild stress. Psychopharmacology (Berl) 1991, 104: 255–259.
Ducottet C, Aubert A, Belzung C: Susceptibility to subchronic unpredictable stress is related to individual reactivity to threat stimuli in mice. Behav Brain Res 2004, 155: 291–299.
Groenink L, Dirks A, Verdouw PM, Schipholt M, Veening JG, van der Gugten J, Olivier B: HPA axis dysregulation in mice overexpressing corticotropin releasing hormone. Biol Psychiatry 2002, 51: 875–881.
Sachar EJ, Hellman L, Roffwarg HP, Halpern FS, Fukushima DK, Gallagher TF: Disrupted 24-hour patterns of cortisol secretion in psychotic depression. Arch Gen Psychiatry 1973, 28: 19–24.
O’Brien JT, Lloyd A, McKeith I, Gholkar A, Ferrier N: A longitudinal study of hippocampal volume, cortisol levels, and cognition in older depressed subjects. Am J Psychiatry 2004, 161: 2081–2090.
Claustrat B, Chazot G, Brun J, Jordan D, Sassolas G: A chronobiological study of melatonin and cortisol secretion in depressed subjects: plasma melatonin, a biochemical marker in major depression. Biol Psychiatry 1984, 19: 1215–1228.
Deuschle M, Schweiger U, Weber B, Gotthardt U, Korner A, Schmider J, Standhardt H, Lammers CH, Heuser I: Diurnal activity and pulsatility of the hypothalamus-pituitary-adrenal system in male depressed patients and healthy controls. J Clin Endocrinol Metab 1997, 82: 234–238.
Surget A, Wang Y, Leman S, Ibarguen-Vargas Y, Edgar N, Griebel G, Belzung C, Sibille E: Corticolimbic transcriptome changes are state-dependent and region-specific in a rodent model of depression and of antidepressant reversal. Neuropsychopharmacology 2009, 34: 1363–1380.
Willner P: Validation criteria for animal models of human mental disorders: learned helplessness as a paradigm case. Prog Neuropsychopharmacol Biol Psychiatry 1986, 10: 677–690.
Berger M, Bossert S, Krieg JC, Dirlich G, Ettmeier W, Schreiber W, von Zerssen D: Interindividual differences in the susceptibility of the cortisol system: an important factor for the degree of hypercortisolism in stress situations? Biol Psychiatry 1987, 22: 1327–1339.
Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ: Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci 2006, 9: 519–525.
Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, Laplant Q, Graham A, Lutter M, Lagace DC, Ghose S, Reister R, Tannous P, Green TA, Neve RL, Chakravarty S, Kumar A, Eisch AJ, Self DW, Lee FS, Tamminga CA, Cooper DC, Gershenfeld HK, Nestler EJ: Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 2007, 131: 391–404.
Shimamoto A, Debold JF, Holly EN, Miczek KA: Blunted accumbal dopamine response to cocaine following chronic social stress in female rats: exploring a link between depression and drug abuse. Psychopharmacology (Berl) 2011, 218: 271–279.
Holly EN, Shimamoto A, Debold JF, Miczek KA: Sex differences in behavioral and neural cross-sensitization and escalated cocaine taking as a result of episodic social defeat stress in rats. Psychopharmacology (Berl) 2012, 224: 179–188.
Bourke CH, Neigh GN: Exposure to repeated maternal aggression induces depressive-like behavior and increases startle in adult female rats. Behav Brain Res 2012, 227: 270–275.
Trainor BC, Pride MC, Villalon Landeros R, Knoblauch NW, Takahashi EY, Silva AL, Crean KK: Sex differences in social interaction behavior following social defeat stress in the monogamous California mouse ( Peromyscus californicus ). PLoS One 2011, 6: e17405.
Trainor BC, Takahashi EY, Campi KL, Florez SA, Greenberg GD, Laman-Maharg A, Laredo SA, Orr VN, Silva AL, Steinman MQ: Sex differences in stress-induced social withdrawal: independence from adult gonadal hormones and inhibition of female phenotype by corncob bedding. Horm Behav 2013, 63: 543–550.
Seligman ME, Beagley G: Learned helplessness in the rat. J Comp Physiol Psychol 1975, 88: 534–541.
Chourbaji S, Zacher C, Sanchis-Segura C, Dormann C, Vollmayr B, Gass P: Learned helplessness: validity and reliability of depressive-like states in mice. Brain Res Brain Res Protoc 2005, 16: 70–78.
Dalla C, Edgecomb C, Whetstone AS, Shors TJ: Females do not express learned helplessness like males do. Neuropsychopharmacology 2008, 33: 1559–1569.
Chourbaji S, Pfeiffer N, Dormann C, Brandwein C, Fradley R, Sheardown M, Gass P: The suitability of 129SvEv mice for studying depressive-like behaviour: both males and females develop learned helplessness. Behav Brain Res 2010, 211: 105–110.
Arnold AP: The organizational-activational hypothesis as the foundation for a unified theory of sexual differentiation of all mammalian tissues. Horm Behav 2009, 55: 570–578.
Arnold AP: Mouse models for evaluating sex chromosome effects that cause sex differences in non-gonadal tissues. J Neuroendocrinol 2009, 21: 377–386.
McCarthy MM, Arnold AP: Reframing sexual differentiation of the brain. Nat Neurosci 2011, 14: 677–683.
McCarthy MM, Wright CL, Schwarz JM: New tricks by an old dogma: mechanisms of the organizational/activational hypothesis of steroid-mediated sexual differentiation of brain and behavior. Horm Behav 2009, 55: 655–665.
Baum MJ: Differentiation of coital behavior in mammals: a comparative analysis. Neurosci Biobehav Rev 1979, 3: 265–284.
Arnold AP, Breedlove SM: Organizational and activational effects of sex steroids on brain and behavior: a reanalysis. Horm Behav 1985, 19: 469–498.
Schulz KM, Molenda-Figueira HA, Sisk CL: Back to the future: the organizational-activational hypothesis adapted to puberty and adolescence. Horm Behav 2009, 55: 597–604.
Arnold AP, Chen X, Itoh Y: What a difference an X or Y makes: sex chromosomes, gene dose, and epigenetics in sexual differentiation. Handb Exp Pharmacol 2012, 259: 2–9.
Jacobs PA, Strong JA: A case of human intersexuality having a possible XXY sex-determining mechanism. Nature 1959, 183: 302–303.
Sinclair AH, Berta P, Palmer MS, Hawkins JR, Griffiths BL, Smith MJ, Foster JW, Frischauf AM, Lovell-Badge R, Goodfellow PN: A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 1990, 346: 240–244.
Koopman P, Gubbay J, Vivian N, Goodfellow P, Lovell-Badge R: Male development of chromosomally female mice transgenic for Sry. Nature 1991, 351: 117–121.
Arnold AP, Chen X: What does the “four core genotypes” mouse model tell us about sex differences in the brain and other tissues? Front Neuroendocrinol 2009, 30: 1–9.
Ikeda Y, Shen WH, Ingraham HA, Parker KL: Developmental expression of mouse steroidogenic factor-1, an essential regulator of the steroid hydroxylases. Mol Endocrinol 1994, 8: 654–662.
Parker KL, Schimmer BP: Steroidogenic factor 1: a key determinant of endocrine development and function. Endocr Rev 1997, 18: 361–377.
Budefeld T, Grgurevic N, Tobet SA, Majdic G: Sex differences in brain developing in the presence or absence of gonads. Dev Neurobiol 2008, 68: 981–995.
Budefeld T, Tobet SA, Majdic G: Gonadal hormone independent sex differences in steroidogenic factor 1 knockout mice brain. Slov Vet Zb 2010, 47: 167–170.
Budefeld T, Tobet SA, Majdic G: Altered position of cell bodies and fibers in the ventromedial region in SF-1 knockout mice. Exp Neurol 2011, 232: 176–184.
Grgurevic N, Budefeld T, Rissman EF, Tobet SA, Majdic G: Aggressive behaviors in adult SF-1 knockout mice that are not exposed to gonadal steroids during development. Behav Neurosci 2008, 122: 876–884.
Grgurevic N, Budefeld T, Spanic T, Tobet SA, Majdic G: Evidence that sex chromosome genes affect sexual differentiation of female sexual behavior. Horm Behav 2012, 61: 719–724.
Arnold AP: Conceptual frameworks and mouse models for studying sex differences in physiology and disease: why compensation changes the game. Exp Neurol 2014, 259: 2–9.
Sequeira A, Mamdani F, Ernst C, Vawter MP, Bunney WE, Lebel V, Rehal S, Klempan T, Gratton A, Benkelfat C, Rouleau GA, Mechawar N, Turecki G: Global brain gene expression analysis links glutamatergic and GABAergic alterations to suicide and major depression. PLoS One 2009, 4: e6585.
Luscher B, Shen Q, Sahir N: The GABAergic deficit hypothesis of major depressive disorder. Mol Psychiatry 2011, 16: 383–406.
Guilloux JP, Douillard-Guilloux G, Kota R, Wang X, Gardier AM, Martinowich K, Tseng GC, Lewis DA, Sibille E: Molecular evidence for BDNF- and GABA-related dysfunctions in the amygdala of female subjects with major depression. Mol Psychiatry 2012, 17: 1130–1142.
Tripp A, Oh H, Guilloux JP, Martinowich K, Lewis DA, Sibille E: BDNF signaling and subgenual anterior cingulate cortex dysfunction in major depression. Am J Psychiatry 2012, 169: 1194–1202.
Sibille E, Morris HM, Kota RS, Lewis DA: GABA-related transcripts in the dorsolateral prefrontal cortex in mood disorders. Int J Neuropsychopharmacol 2011, 14: 721–734.
Seney ML, Chang LC, Oh H, Wang X, Tseng GC, Lewis DA, Sibille E: The role of genetic sex in affect regulation and expression of GABA-related genes across species. Front Psychiatry 2013, 4: 104.
Guilloux JP, Seney M, Edgar N, Sibille E: Integrated behavioral z-scoring increases the sensitivity and reliability of behavioral phenotyping in mice: relevance to emotionality and sex. J Neurosci Methods 2011, 197: 21–31.
Hisasue S, Seney ML, Immerman E, Forger NG: Control of cell number in the bed nucleus of the stria terminalis of mice: role of testosterone metabolites and estrogen receptor subtypes. J Sex Med 2010, 7: 1401–1409.
Murray EK, Hien A, de Vries GJ, Forger NG: Epigenetic control of sexual differentiation of the bed nucleus of the stria terminalis. Endocrinology 2009, 150: 4241–4247.
del Abril A, Segovia S, Guillamon A: The bed nucleus of the stria terminalis in the rat: regional sex differences controlled by gonadal steroids early after birth. Brain Res 1987, 429: 295–300.
Guillamon A, Segovia S, del Abril A: Early effects of gonadal steroids on the neuron number in the medial posterior region and the lateral division of the bed nucleus of the stria terminalis in the rat. Brain Res Dev Brain Res 1988, 44: 281–290.
Goel N, Bale TL: Organizational and activational effects of testosterone on masculinization of female physiological and behavioral stress responses. Endocrinology 2008, 149: 6399–6405.
Viau V, Meaney MJ: Variations in the hypothalamic-pituitary-adrenal response to stress during the estrous cycle in the rat. Endocrinology 1991, 129: 2503–2511.
Laplant Q, Chakravarty S, Vialou V, Mukherjee S, Koo JW, Kalahasti G, Bradbury KR, Taylor SV, Maze I, Kumar A, Graham A, Birnbaum SG, Krishnan V, Truong HT, Neve RL, Nestler EJ, Russo SJ: Role of nuclear factor kappaB in ovarian hormone-mediated stress hypersensitivity in female mice. Biol Psychiatry 2009, 65: 874–880.
Frye CA, Seliga AM: Testosterone increases analgesia, anxiolysis, and cognitive performance of male rats. Cogn Affect Behav Neurosci 2001, 1: 371–381.
De Vries GJ: Minireview: sex differences in adult and developing brains: compensation, compensation, compensation. Endocrinology 2004, 145: 1063–1068.
Palaszynski KM, Smith DL, Kamrava S, Burgoyne PS, Arnold AP, Voskuhl RR: A yin-yang effect between sex chromosome complement and sex hormones on the immune response. Endocrinology 2005, 146: 3280–3285.
Bonthuis PJ, Cox KH, Rissman EF: X-chromosome dosage affects male sexual behavior. Horm Behav 2012, 61: 565–572.
Choudary PV, Molnar M, Evans SJ, Tomita H, Li JZ, Vawter MP, Myers RM, Bunney WE Jr, Akil H, Watson SJ, Jones EG: Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc Natl Acad Sci U S A 2005, 102: 15653–15658.
Sanacora G, Gueorguieva R, Epperson CN, Wu YT, Appel M, Rothman DL, Krystal JH, Mason GF: Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression. Arch Gen Psychiatry 2004, 61: 705–713.
Sanacora G, Mason GF, Rothman DL, Behar KL, Hyder F, Petroff OA, Berman RM, Charney DS, Krystal JH: Reduced cortical gamma-aminobutyric acid levels in depressed patients determined by proton magnetic resonance spectroscopy. Arch Gen Psychiatry 1999, 56: 1043–1047.
Gabbay V, Mao X, Klein RG, Ely BA, Babb JS, Panzer AM, Alonso CM, Shungu DC: Anterior cingulate cortex gamma-aminobutyric acid in depressed adolescents: relationship to anhedonia. Arch Gen Psychiatry 2012, 69: 139–149.
Tripp A, Kota RS, Lewis DA, Sibille E: Reduced somatostatin in subgenual anterior cingulate cortex in major depression. Neurobiol Dis 2011, 42: 116–124.
Cowen PJ, Parry-Billings M, Newsholme EA: Decreased plasma tryptophan levels in major depression. J Affect Disord 1989, 16: 27–31.
Asberg M, Thoren P, Traskman L, Bertilsson L, Ringberger V: “Serotonin depression”—a biochemical subgroup within the affective disorders? Science 1976, 191: 478–480.
Arango V, Underwood MD, Mann JJ: Serotonin brain circuits involved in major depression and suicide. Prog Brain Res 2002, 136: 443–453.
Richardson-Jones JW, Craige CP, Guiard BP, Stephen A, Metzger KL, Kung HF, Gardier AM, Dranovsky A, David DJ, Beck SG, Hen R, Leonardo ED: 5-HT1A autoreceptor levels determine vulnerability to stress and response to antidepressants. Neuron 2010, 65: 40–52.
Richardson-Jones JW, Craige CP, Nguyen TH, Kung HF, Gardier AM, Dranovsky A, David DJ, Guiard BP, Beck SG, Hen R, Leonardo ED: Serotonin-1A autoreceptors are necessary and sufficient for the normal formation of circuits underlying innate anxiety. J Neurosci 2011, 31: 6008–6018.
Suhara T, Nakayama K, Inoue O, Fukuda H, Shimizu M, Mori A, Tateno Y: D1 dopamine receptor binding in mood disorders measured by positron emission tomography. Psychopharmacology (Berl) 1992, 106: 14–18.
Dougherty DD, Bonab AA, Ottowitz WE, Livni E, Alpert NM, Rauch SL, Fava M, Fischman AJ: Decreased striatal D1 binding as measured using PET and [11C] SCH 23,390 in patients with major depression with anger attacks. Depress Anxiety 2006, 23: 175–177.
Cannon DM, Klaver JM, Peck SA, Rallis-Voak D, Erickson K, Drevets WC: Dopamine type-1 receptor binding in major depressive disorder assessed using positron emission tomography and [11C] NNC-112. Neuropsychopharmacology 2009, 34: 1277–1287.
Tye KM, Mirzabekov JJ, Warden MR, Ferenczi EA, Tsai HC, Finkelstein J, Kim SY, Adhikari A, Thompson KR, Andalman AS, Gunaydin LA, Witten IB, Deisseroth K: Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature 2013, 493: 537–541.
Seney ML, Ekong KI, Ding Y, Tseng GC, Sibille E: Sex chromosome complement regulates expression of mood-related genes. Biol Sex Differ 2013, 4: 20.
Acknowledgements
This work was supported by the National Institute of Mental Health MH084060 (ES), MH085111 (ES), and MH103473 (MLS). Marianne Seney in 2014 served as a consultant for NeuroPhage Pharmaceuticals, providing technique-based support.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Both authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
Cite this article
Seney, M.L., Sibille, E. Sex differences in mood disorders: perspectives from humans and rodent models. Biol Sex Differ 5, 17 (2014). https://doi.org/10.1186/s13293-014-0017-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13293-014-0017-3