
Abstract
Rapid technological development has created an urgent need for improved evaluation of algorithms for the analysis of cancer genomics data. We outline how challenge-based assessment may help fill this gap by leveraging crowd-sourcing to distribute effort and reduce bias.
Similar content being viewed by others


Computational biology comprises three inter-connected activities: algorithm development, validation through benchmarking, and application. In the biomedical sciences, benchmarking occupies a central and indispensable role as it maps algorithms from the space of theoretical possibilities to the realm of practical value. Critically, this process attributes specific probabilities to an algorithm’s discovery of biologically relevant knowledge (measured by the sensitivity of the algorithm) while not overwhelming the researcher with incorrect predictions (quantified by the algorithm specificity). Benchmarking is, however, a complex task, requiring the creation of comprehensive gold standards and the design of sophisticated validation strategies that may require additional experimental data. Indeed, as the use of computational methods in biomedical research becomes widespread, the need for appropriate benchmarking projects, especially those involving community participation, is substantially growing (Table 1). In particular, the rapidly increasing size of whole-genome molecular profile datasets from large sample repositories underscores the importance of benchmarking; it has become virtually impossible to validate algorithmic predictions that are based on such large datasets systematically.
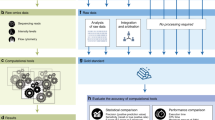
Benchmarking is not a matter of simply running a few algorithms on a few datasets and comparing the results. Drawing generalizable conclusions from the exercise requires significant care in design and execution. The maturity of bioinformatics as a discipline has been greatly advanced by the adoption of key principles that guide robust method evaluation, including evaluator objectiveness (lack of bias), clearly defined scoring metrics that align with real-world goals, and the public release of gold-standard datasets and of the results and code of prediction algorithms. Challenge-based (also known as `competition-based’) method assessment is an increasingly popular mechanism for benchmarking [1],[2]. In this type of study an impartial group of scientists organizes a `challenge’ that is based on a carefully curated dataset. This dataset is typically split into a training dataset, a validation dataset (which might be used in real-time leaderboards, typically implemented as a table that reports the comparative performance of the methods under development), and a gold standard (or test) dataset that is withheld from challenge participants and used for final evaluation (Figure 1). Following algorithm development on the training dataset and real-time feedback to participants based on the validation dataset and reported in the leaderboard, the challenge organizers can objectively evaluate the quality of final submitted predictions using a gold-standard dataset. Such a design closely reflects the actual difficulties faced by real-world users trying to determine whether an algorithm generalizes to unseen cases.

Typical design of a crowd-sourced challenge. A dataset is split into a training set, a validation (or leaderboard set) and the test set (or gold standard). Participants have access to the challenge input data and the known answers for just the training set. For the validation and test sets only, the challenge input data are provided but the answers to the challenge questions are withheld. In the challenge open phase, participants optimize their algorithms by making repeated submissions to predict the validation set answers. These submissions are scored and returned to the participants who can use the information to improve their methods. In the final evaluation phase, the optimized algorithms are submitted and evaluated against the final test set (the gold standard), and the resulting scores are used to compute the statistical significance and the ranking of the participating algorithms.
When flawed, benchmarking can lead to the emergence of suboptimal standards that may be applied to many large datasets, imposing an immense cost to the community and creating misleading results. Conversely, the acceptance of knowledge without robust benchmarking can lead to the adoption of inaccurate conventions. For example, during the 1990s, it was generally accepted that the number of loci coding for proteins in the human genome was 100,000, a number that was based on unverified hypotheses [3]. When the human genome was finally sequenced in 2000, the total number of coding loci was found to be a factor of 5 lower. Similarly, a design error in the early implementation of the GC Robust Multi-Array (GCRMA) algorithm, which was revealed by systematic benchmarking of network reconstruction analyses, may have led to the publication of thousands of papers that contain incorrect mRNA abundance profiles before the error was detected and corrected [4]. As a third example, in 2006, a group of Duke University researchers published a pair of high-impact papers claiming accurate prediction of the prognosis of lung cancer patients and of chemotherapy-sensitivity in lung, breast and ovarian cancers. Attempts to reproduce those claims ensued almost immediately, with most of the results falling short of replication because of a combination of programming and data-entry errors, and possible fraud [5]. Proper objective benchmarking by a neutral third-party on private validation data helps to resolve quickly or to detect many of the issues associated with these kinds of studies.
One concern in algorithm benchmarking and validation is that computational biology algorithms are often developed and evaluated by the same researchers. This creates an inherent conflict of interest, where objective assessment of accuracy is polluted by the fact that the developers become simultaneously judge, jury and executioner of the validity of their own work. This can result in biases in study design and over-optimistic performance estimates, whether intentional or unintentional [6]. For instance, the use of non-blinded data in the evaluation by methods developers of their own protein structure prediction methods led, in the early’ 80s, to the false belief that protein structure prediction was essentially a solved problem. Not until 1994, when double-blinded data were used in the first Workshop on the Critical Assessment of Protein Structure Prediction (CASP), was a very different picture revealed [7].
Challenge-based benchmarking efforts, such as CASP [8]-[10], CAFA [11] and DREAM [12],[13], among others (Table 1), offer a robust framework for algorithm evaluation. These efforts have proven the value of engaging both active challenge leaders and motivated algorithm developers to improve their work in a forum that has high visibility and rapid feedback.
We believe that challenge-based methods assessment will play an increasingly important role in standardizing and optimizing the analysis of cancer genomics data, and its broader adoption will drive progress in both algorithm development and biological discovery. Conversely, failing to exploit challenge-benchmarking as a fundamental validation methodology for cancer genomics algorithms may result in lost opportunities to translate results derived from best-in-class methods into patient care.
Here, we provide our perspective on the growing use of challenge-based methods to benchmark algorithms in cancer genomics. We outline the different types of problems faced and some of the key considerations that need to be explored to determine whether a challenge might be successful, and to provide suggestions for challenge organization and execution. Finally, we look to the future and consider how challenge-based assessment may change in the coming decade.
Challenge design and dynamics
Over the past few years, an established challenge-based design paradigm has emerged in which portions of a private (that is, not globally released) dataset are made publicly available according to a predefined schedule. Such a dataset provides increased user engagement based on continuous feedback; an opportunity for participants to refine and improve their methods on the basis of results obtained throughout the challenge; and multiple independent rounds of validation, which can be used to assess the consistency and robustness of results. After the initial training dataset is made publicly available, a real-time leaderboard can be generated in which the performance of different algorithms is evaluated against a withheld private portion of the data (Figure 1). Previous research has shown that the provision of real-time feedback is among the most important factors in ensuring user engagement in crowd-sourcing projects [14]. (Here, we use the term crowd-sourcing in the sense that a community of tens to hundreds of researchers are engaged in working on the same problem. In other contexts, crowd-sourcing activities may engage different numbers of participants.) After a period of time in which several iterations of the leaderboard can be posted, one of the participating groups is declared the best performer in this initial phase of the challenge, either on the basis of their position on the leaderboard or because they were the first to reach some pre-specified performance level. The challenge may include multiple rounds of assessment based on different portions of the private data. A final round is typically invoked in which methods are rated against a withheld evaluation dataset to determine the overall challenge winner (Figure 1). The most robust validation set is often reserved for this final evaluation - often with larger sample size, newly generated data or prospective validation designed on the basis of challenge results. Each participating team submits a small number (for example, one to five) of independent predictions made by their algorithm(s), which are scored and ranked to determine a winner. Finally, the public release of all of the data kept private throughout the challenge, along with the predictions and ideally source code from each group, provides a long-term resource to spur further development of new and improved methods.
The collection of algorithm source code allows developers to share insights that promote future improvements. If required as part of the final submission, this code can also be used to ensure objective scoring and verification of reproducibility. In the 2012 Sage Bionetworks-DREAM Breast Cancer Prognosis Challenge, participants were required to submit their models as open source R-code [15] that was visible to all participants and executed by an automated system to generate the results reported on the leaderboard. This was enabled by Synapse [16], a software platform that supports scientific challenges as well as large distributed collaborations, such as those in the TCGA Pan-Cancer consortium [17]. Planned challenges, such as the RNA-seq follow-up to the ICGC-TCGA DREAM Somatic Mutation Calling (SMC) Challenge, are considering the use of cloud-computing solutions to provide a central computing facility and a harness upon which contestant code is directly run. This will inherently force the deposition of complete analysis workflows, which can be run routinely on new datasets. Further, this approach would help to standardize application programming interfaces and file formats, such that multiple algorithms use similar inputs and produce easily comparable outputs. This vision of interoperability is shared by many practitioners in the field and has most recently been championed by the Global Alliance for Genomics And Health [18].
Several criteria should be used to help participants limit over-fitting to the training data. Over-fitting is a known peril in statistics, occurring when a predictive model has enough flexibility in its parameters that optimization effectively leads to `memorization’ of the training data and an inability to generalize to unseen cases. The most common way to help participants avoid over-fitting, while enabling the testing of their models, is to provide leaderboard scoring that is based on a subset of the private dataset, optimally a subset that is not used in the final evaluation. The latter condition is sometimes not feasible (for example, when the number of patients available to predict clinical outcomes is limited), in which case the leaderboard will be based on data that are also used for the final scoring. If this is the case, limiting the number of submissions can reduce over-fitting.
While most challenges share some common design principles, each research area has its own unique characteristics that require customized experimental designs and consideration of risks and benefits. Indeed, the utility of organizing a challenge to help advance a particular research area depends on a balance between challenge-based benchmarking advantages and limitations, as well as consideration of the potential barriers for participation (Table 2). In the sections below, we highlight three research areas in which rapid development of new algorithms has led to a concomitant need for benchmarking: accurate identification of tumor-specific genomic alterations, association of clinical data with genomic profiles (that is, biomarkers) and identifying network-biology features that underlie cancer phenotypes.
Analyzing genome assembly and structural variants
Technologies for identifying cancer-related somatic alterations from genomic or transcriptomic data are advancing extremely rapidly. In only 6 years, next-generation sequencing (NGS) has rapidly progressed from the measurement of millions of short sequences (of around 25 bp) to that of hundreds of millions of longer segments (of around 100 bp). This creates an urgent need for on-going benchmarking studies as old algorithms become rapidly out-dated and new algorithmic approaches are required to handle new technologies and new scales of data. Small-scale studies have resulted in dramatic discordance when different researchers apply their algorithms to the same genomic data (Figure 2) [19]-[21]. These studies have shown that accuracy and generalizability vary dramatically across samples and regions of the genome. The constantly shifting landscape presented by rapidly evolving technologies and tools fuels the urgency in the need to identify the best-performing methods objectively and to re-evaluate them frequently, and to identify particularly error-prone aspects of existing tumor genome analysis methods [22]. Several non-cancer-focused challenge-based benchmarking efforts are on-going, including the Assemblathon benchmarking of de novo sequence assembly algorithms [23] and the CLARITY Challenge for standardizing clinical genome sequencing analysis and reporting [24] (Table 1).

Different researchers studying the same data may arrive at discordant conclusions. Benchmarking becomes essential as a way to separate true findings from spurious ones. (Illustration by Natasha Stolovitzky-Brunner© inspired by the parable of the six blind men and the elephant).
Challenge-based benchmarking of methods for somatic variant detection in cancer faces several unique hurdles. First, genomic sequence is inherently identifiable [25], and is thus considered personal health information (PHI) in many countries. This places a burden on challenge contestants to acquire ethics approval from the appropriate authorities, such as dbGaP in the USA or ICGC in Canada. Second, because of the inherent complexity of both the data and file formats, it may be difficult for researchers from other fields to acquire sufficient domain knowledge to compete effectively against domain experts. This point may be ameliorated by gamifying the problem, that is, using game tools that require puzzle solving or geometric thinking to engage users in genomics problems [26],[27]. Gamification may not be possible or appropriate, however, because it may require sacrificing domain-specific prior knowledge that is essential to the correct solution. Third, the size of the raw genomic data necessary to perform these challenges creates a `big-data’ problem. For example, the ICGC-TCGA DREAM SMC Challenge [28] (Table 1) involved transmitting over 10 TB of data to every contestant, so that each had a copy of the 15 tumor-normal whole-genome pairs. Two different solutions to this problem are to provide access to high-speed, cloud-based download technologies (such as GeneTorrent or Aspera) or to provide co-location of computers and data in a hosted environment [29]. The latter solution has the advantage of providing implementations of the best-performing algorithms in a form that is more readily redistributed to the community, as well as allowing more `democratized’ participation for groups that do not have large in-house computing resources. Nevertheless, this solution also has disadvantages: cloud-computing may require additional overhead expenditure for groups that are familiar with developing methods within their local computing environments; many researchers have access to in-house computing options subsidized by their institution and have limited incentive to transfer their analysis to the cloud; and access permissions for some datasets can hinder redistribution through cloud platforms. Furthermore, the assessment of predictions is challenging because the ground-truth for genetic alterations is unknown. The SMC Challenge employs two strategies for evaluation. The first involves an in silico method for simulating cancer genomes called BAMSurgeon, which was developed to allow the comparison of methods predictions against a synthetic ground-truth (work by Ewing and colleagues). In the second strategy, targeted deep-sequencing allows prospective validation of a large number of predicted mutations, chosen by an algorithm that most accurately computes false-positive and false-negative rates across submissions. It is unclear how important it is for prospective validation data to be orthogonal to that used by the original challenge participants. Verification in TCGA projects typically relies on deep sequencing using the same technology, but on selected targets and with the construction of new sequencing libraries. This approach assumes that most errors are randomly distributed and/or associated with only a small fraction of reads. The more orthogonal the validation technology, the more this assumption is relaxed. Nevertheless, the error profile of the final evaluation dataset is crucial, and there are currently no error-free approaches to generating this gold-standard data for NGS.
Finding genomic biomarkers that are associated with phenotype
Once a set of somatic variants have been identified from genomic interrogation of patient-derived samples, one of the most common analyses is to attempt to develop biomarkers that can predict patient survival, response to therapy or other outcomes [30]-[33]. The development of genomic-based personalized medicine has immense clinical potential, but the optimal approach to predicting such biomarkers de novo remains poorly understood and controversial. Indeed, it is widely known that inferred biomarkers are highly sensitive to factors such as choice of algorithm and data pre-processing methods [34]-[37].
Nevertheless, developing challenges to benchmark biomarker discovery problems is relatively straightforward. Participants are given training data in which features (for example, genome-wide mRNA transcript abundance) are paired with outcome (for example, patient survival) data. Participants are given only the features for the test set and asked to predict the outcome data using a model inferred from the training data. Alternatively, participants may submit trained models as executable code to be run on the test data, thus allowing the test feature data to be hidden from participants [15]. Model results are scored on the basis of the correspondence between predicted and measured outcome data from the test set.
Prediction challenges have been employed in many domains outside of biomedical research [38]. Because biomarker-based challenges fit the setup of the classic supervised machine-learning paradigm, they attract new ideas and participation from the broader machine-learning community. Benchmarking in biomarker discovery is crucial, however, as outlined by the case of the retracted Duke study on chemotherapy selection noted above.
Two key difficulties exist in the creation of benchmarking challenges for biomarker discovery. First, the ideal datasets for biomarker-discovery challenges are uniquely defined, especially when data were collected from large cohorts requiring long-term follow-up or expensive standardized treatment protocols (such as clinical trials). These datasets can potentially lead to high-impact publications or concerns over the intellectual property of the data-generating groups. Second, the potential size of patient cohorts is currently limiting for many biomarker-development questions. If the amount of data available is inadequate, they may not generate enough statistical power to distinguish the performance of the top-ranked groups accurately. These factors also complicate the ability to obtain independent datasets for final method assessment. Despite these problems, several successful challenges pertaining to diagnostics, prognostics and treatment outcomes have been conducted, including the MAQC-II study [39], the IMPROVER Challenge on Diagnostic Signatures [40], the Sage Bionetworks DREAM Breast Cancer Prognostics Challenge [15], and the DREAM AML Treatment Outcome Challenge [41].
Inferring biological networks underlying cancer phenotypes
Identifying the relationships between biological (transcriptional and signaling) networks and cancer onset and progression is another potential area for challenge benchmarking. Network analysis involves several aspects, including the coherent modeling of different types of alteration and dysregulation events and their integration into a unified network-based model [42]-[44]. One of the major problems with organizing challenges in this area is that the underlying cellular regulatory networks are mostly unknown, especially in complex systems such mammalian tumor cells. So how can a challenge be organized when a pre-known gold-standard network cannot be defined? Several strategies employed by the DREAM project include using synthetic biology networks [13], in silico networks [45], and experimentally assessed bacterial networks [46]. An alternative strategy is to evaluate methods on the basis of their ability to predict the response of a system to a set of perturbations, such as drugs or receptor ligands, as surrogates for predicting the underlying network connectivity [47]. The introduction of ingenious surrogates to the gold standard has enabled the formulation of other network reverse-engineering challenges, such as the 2013 HPN-DREAM Breast Cancer Network Inference Challenge [48]. In this challenge, participants were asked to submit predicted signaling networks that were activated by a set of stimuli in four breast cancer cell lines. These networks were scored on the basis of their ability to identify the set of proteins that are downstream of a given phosphoprotein. The predicted protein set was compared to an experimentally determined set of proteins (the surrogate gold standard), defined as those proteins whose phosphorylation levels were affected by inhibiting that phosphoprotein. Further research on benchmarking network-inference algorithms would be highly beneficial to help advance the field of network biology, whose role in unraveling biological mechanisms in cancer is hard to overestimate.
The truth is hard to find
From the previous discussion, it is clear that the single most crucial aspect in benchmarking is the definition and assembly of gold standards. A gold standard fundamentally defines the problem under study, and it provides the limiting resolution of error for the overall endeavor. As outlined in this article, gold standards can be defined in several ways. First, a single experiment can be performed with portions of the resulting data used for training and evaluation. This approach avoids experimental inconsistencies, but requires that a large selection of true results is generated prior to the challenge. Simulated datasets are ideal for this strategy but have been criticized as only partially representing a biological system [49]. While validation of simulated data is straight forward, because the ground-truth is completely known, in most cases the value of benchmarking is perceived to be in the ability to assess best-performing methods when applied to real biological data as opposed to simulated data. An important caveat is that the synthetic data may fail to reflect some of the underlying assumptions of the system they attempt to emulate. Indeed, the most common question about simulations is how well they reflect experimental samples [49].
Second, for systems that are difficult to benchmark directly, such as the structure of a biological network, characteristics of the systems can be evaluated instead. These might include the effects of the systems’ perturbation or other phenomena, such as the identification of the networks that best predict patient outcomes.
Third, the results of a study can be validated after the challenge is completed by additional experimental work, either on the same sample or on others. This has the advantage of directly addressing the predictions made by challenge participants, but has the disadvantage of introducing a time lag between challenge completion and the availability of full results. In addition, the effort and cost of follow-up validation may be prohibitive given the resources available to the challenge organizers.
For genomic studies, wet-lab validation can be both time-consuming and expensive. For example, the MAQC study considered approximately 20,000 genes on microarray platforms, but only validated approximately 1,000 (5%) by real-time PCR as a gold standard [50]. Because of this cost, both in terms of time and money, it is critical that a good validation be sufficiently representative, providing similar levels of statistical power for assessing the accuracy of each group. In the context of somatic mutation calling, this means selecting calls that are unique to individual predictors as well as those common to multiple predictors. Indeed, the validation techniques will often be experimentally limited to a subset of results, leaving a bias in the distribution of what is tested. There is thus a clear need for research into the optimal selection of validation candidates in many biological settings. Further, validating a small subset (<10%) of results comes with the possibility, however small, of producing an incorrect relative ordering of different algorithms. In practice, a combination of synthetic and real-world validation is best, and finding the right balance is challenge-dependent.
Finally, some very important elements of cancer genomics are difficult to validate. For example, almost all NGS analyses rely on sequence alignment as a first step. It is, however, very difficult to benchmark the accuracy of an alignment algorithm on real tumor data, because there is no obvious way to create a ground-truth dataset. Thus, rather than benchmarking the aligners, challenges benchmark the results of entire pipelines such as those for detecting somatic variants [28], which may incorporate different aligners and different data pre-processing and statistical approaches. Similarly, it is of great interest to infer cancer-driver genes. Unfortunately, the definition of a `driver gene’ (beyond simple statistical recurrence) is unclear, and does not yet allow unambiguous, high-throughput experimental validation. Most experimental techniques in this area probe only one aspect of a driver gene (such as its influence on proliferation or metastasis), while many subtle phenotypes (such as angiogenesis or local spread) are challenging to probe. Also, these designs ignore the potentially polygenic nature of tumor initiation and progression. In designing a new challenge, one of the first questions must be whether or not suitable gold-standard test datasets can be generated.
Closing considerations
Benchmarking is a fundamental part of computational biology and is increasingly being appreciated by the biomedical community as a whole. Recent benchmarking studies both within [19],[51] and outside of cancer genomics [39],[52]-[54] have helped highlight new ways to analyze data and have prompted reconsideration of the error profiles of datasets. Challenge-based assessments have also recently surged in other fields [55] in which the use of incentives (including prizes and prestige) have stimulated increased attention and algorithm development [56].
As the profile of the results of benchmarking studies increases, it is becoming increasingly clear that benchmarking itself is a serious scientific endeavor. The design of a challenge is non-trivial and in some ways is easy `to get wrong’ - there needs to be a careful integration between experts in challenge-based benchmarking and domain experts in the challenge topic. At the outset, there is a fundamental requirement for the benchmarking team to foster a community that supports and promotes the exercise. Indeed, some topic areas may be unsuitable to challenge-based benchmarking because a sufficiently big community of interested algorithm developers has not yet emerged (although in these cases, appropriate incentives may be useful in helping to focus attention on a potential challenge topic). Further, the challenge organizing team must be able to assure the broader community of its neutrality and objectivity. There is a clear advantage to building groups of `challenge-based benchmarking experts’ who can bring their expertise to diverse topics within cancer genomics, or any other field. Such groups may be well-placed to develop and optimize the statistical methods needed to improve challenge-based benchmarks. Several groups are developing the expertise to facilitate this process, including CASP, DREAM, CAFA and others (Table 1).
Cancer genomics is characterized by rapid technological development, and this trend is likely to persist for many years. As a result, benchmarking cannot be a static endeavor. Rather, each new technology will have its own specific error profiles and distinct algorithms that are used for data analysis. In a world of continual technological and algorithmic innovation, it may be impossible to have definitive, permanent benchmarks, because any effort will be based on a snapshot of technology and will rapidly become out-dated. Instead, a long-running series of `living benchmarks’ may allow the co-evolution of benchmarks with technology. In this mutualistic scenario, regular releases of new datasets capturing the current state of experimental methodologies will allow users at any point in time to identify the best tool for their dataset, and algorithm developers to have a dataset suitable for developing and optimizing methods on the latest data.
Abbreviations
- CASP:
-
Critical Assessment of Protein Structure Prediction
- GCRMA:
-
GC Robust Multi-Array
- PHI:
-
Personal health information
- NGS:
-
Next-generation sequencing
- SMC:
-
Somatic Mutation Calling
References
Costello JC, Stolovitzky G: Seeking the wisdom of crowds through challenge-based competitions in biomedical research. Clin Pharmacol Ther. 2013, 93: 396-398. 10.1038/clpt.2013.36.
Meyer P, Alexopoulos LG, Bonk T, Califano A, Cho CR, de la Fuente A, de Graaf D, Hartemink AJ, Hoeng J, Ivanov NV, Koeppl H, Linding R, Marbach D, Norel R, Peitsch MC, Rice JJ, Royyuru A, Schacherer F, Sprengel J, Stolle K, Vitkup D, Stolovitzky G: Verification of systems biology research in the age of collaborative competition. Nat Biotechnol. 2011, 29: 811-815. 10.1038/nbt.1968.
Pertea M, Salzberg SL: Between a chicken and a grape: estimating the number of human genes. Genome Biol. 2010, 11: 206-10.1186/gb-2010-11-5-206.
Lim WK, Wang K, Lefebvre C, Califano A: Comparative analysis of microarray normalization procedures: effects on reverse engineering gene networks. Bioinformatics. 2007, 23: i282-i288. 10.1093/bioinformatics/btm201.
Baggerly KA, Coombes KR: Deriving chemosensitivity from cell lines: forensic bioinformatics and reproducible research in high-throughput biology. Ann Appl Stat. 2009, 3: 1309-1334. 10.1214/09-AOAS291.
Norel R, Rice JJ, Stolovitzky G: The self-assessment trap: can we all be better than average?. Mol Syst Biol. 2011, 7: 537-10.1038/msb.2011.70.
Moult J, Pedersen JT, Judson R, Fidelis K: A large-scale experiment to assess protein structure prediction methods. Proteins. 1995, 23: ii-v. 10.1002/prot.340230303.
Cozzetto D, Kryshtafovych A, Tramontano A: Evaluation of CASP8 model quality predictions. Proteins. 2009, 77: 157-166. 10.1002/prot.22534.
Shi S, Pei J, Sadreyev RI, Kinch LN, Majumdar I, Tong J, Cheng H, Kim BH, Grishin NV: Analysis of CASP8 targets, predictions and assessment methods. Database (Oxford). 2009, 2009: bap003-10.1093/database/bap003.
Tramontano A, Morea V: Assessment of homology-based predictions in CASP5. Proteins. 2004, 55: 782-10.1002/prot.20187.
Radivojac P, Clark WT, Oron TR, Schnoes AM, Wittkop T, Sokolov A, Graim K, Funk C, Verspoor K, Ben-Hur A, Pandey G, Yunes JM, Talwalkar AS, Repo S, Souza ML, Piovesan D, Casadio R, Wang Z, Cheng J, Fang H, Gough J, Koskinen P, Törönen P, Nokso-Koivisto J, Holm L, Cozzetto D, Buchan DW, Bryson K, Jones DT, Limaye B, et al: A large-scale evaluation of computational protein function prediction. Nat Methods. 2013, 10: 221-227. 10.1038/nmeth.2340.
Prill RJ, Marbach D, Saez-Rodriguez J, Sorger PK, Alexopoulos LG, Xue X, Clarke ND, Altan-Bonnet G, Stolovitzky G: Towards a rigorous assessment of systems biology models: the DREAM3 challenges. PLoS One. 2010, 5: e9202-10.1371/journal.pone.0009202.
Stolovitzky G, Prill RJ, Califano A: Lessons from the DREAM2 challenges. Ann N Y Acad Sci. 2009, 1158: 159-195. 10.1111/j.1749-6632.2009.04497.x.
Athanasopoulos G, Hyndman RJ: The value of feedback in forecasting competitions. Int J Forecast. 2011, 27: 845-849. 10.1016/j.ijforecast.2011.03.002.
Margolin AA, Bilal E, Huang E, Norman TC, Ottestad L, Mecham BH, Sauerwine B, Kellen MR, Mangravite LM, Furia MD, Vollan HK, Rueda OM, Guinney J, Deflaux NA, Hoff B, Schildwachter X, Russnes HG, Park D, Vang VO, Pirtle T, Youseff L, Citro C, Curtis C, Kristensen VN, Hellerstein J, Friend SH, Stolovitzky G, Aparicio S, Caldas C, Børresen-Dale AL: Systematic analysis of challenge-driven improvements in molecular prognostic models for breast cancer. Sci Transl Med. 2013, 5: 181re181-
Synapse; [http://www.sagebase.org/synapse]
Omberg L, Ellrott K, Yuan Y, Kandoth C, Wong C, Kellen MR, Friend SH, Stuart J, Liang H, Margolin AA: Enabling transparent and collaborative computational analysis of 12 tumor types within The Cancer Genome Atlas. Nat Genet. 2013, 45: 1121-1126. 10.1038/ng.2761.
Global Alliance for Genomics and Health ; [http://genomicsandhealth.org]
Kim SY, Speed TP: Comparing somatic mutation-callers: beyond Venn diagrams. BMC Bioinformatics. 2013, 14: 189-10.1186/1471-2105-14-189.
O’Rawe J, Jiang T, Sun G, Wu Y, Wang W, Hu J, Bodily P, Tian L, Hakonarson H, Johnson WE, Wei Z, Wang K, Lyon GJ: Low concordance of multiple variant-calling pipelines: practical implications for exome and genome sequencing. Genome Med. 2013, 5: 28-10.1186/gm432.
Alkan C, Coe BP, Eichler EE: Genome structural variation discovery and genotyping. Nat Rev Genet. 2011, 12: 363-376. 10.1038/nrg2958.
Taking pan-cancer analysis global.Nat Genet 2013, 45:1263.,
Bradnam KR, Fass JN, Alexandrov A, Baranay P, Bechner M, Birol I, Boisvert S, Chapman JA, Chapuis G, Chikhi R, Chitsaz H, Chou WC, Corbeil J, Del Fabbro C, Docking TR, Durbin R, Earl D, Emrich S, Fedotov P, Fonseca NA, Ganapathy G, Gibbs RA, Gnerre S, Godzaridis E, Goldstein S, Haimel M, Hall G, Haussler D, Hiatt JB, Ho IY, et al: Assemblathon 2: evaluating de novo methods of genome assembly in three vertebrate species. Gigascience. 2013, 2: 10-10.1186/2047-217X-2-10.
Brownstein CA, Beggs AH, Homer N, Merriman B, Yu TW, Flannery KC, Dechene ET, Towne MC, Savage SK, Price EN, Holm IA, Luquette LJ, Lyon E, Majzoub J, Neupert P, McCallie D, Szolovits P, Willard HF, Mendelsohn NJ, Temme R, Finkel RS, Yum SW, Medne L, Sunyaev SR, Adzhubey I, Cassa CA, de Bakker PI, Duzkale H, Dworzy Ski P, Fairbrother W, et al: An international effort towards developing standards for best practices in analysis, interpretation and reporting of clinical genome sequencing results in the CLARITY challenge. Genome Biol. 2014, 15: R53-10.1186/gb-2014-15-3-r53.
Gymrek M, McGuire AL, Golan D, Halperin E, Erlich Y: Identifying personal genomes by surname inference. Science. 2013, 339: 321-324. 10.1126/science.1229566.
Good BM, Su AI: Games with a scientific purpose. Genome Biol. 2011, 12: 135-10.1186/gb-2011-12-12-135.
Lee J, Kladwang W, Lee M, Cantu D, Azizyan M, Kim H, Limpaecher A, Yoon S, Treuille A, Das R, Ete RNAP: RNA design rules from a massive open laboratory. Proc Natl Acad Sci U S A. 2014, 111: 2122-2127. 10.1073/pnas.1313039111.
Boutros PC, Ewing AD, Ellrott K, Norman TC, Dang KK, Hu Y, Kellen MR, Suver C, Bare JC, Stein LD, Spellman PT, Stolovitzky G, Friend SH, Margolin AA, Stuart JM: Global optimization of somatic variant identification in cancer genomes with a global community challenge. Nat Genet. 2014, 46: 318-319. 10.1038/ng.2932.
Dudley JT, Butte AJ: In silico research in the era of cloud computing. Nat Biotechnol. 2010, 28: 1181-1185. 10.1038/nbt1110-1181.
Lambin P, van Stiphout RG, Starmans MH, Rios-Velazquez E, Nalbantov G, Aerts HJ, Roelofs E, van Elmpt W, Boutros PC, Granone P, Valentini V, Begg AC, De Ruysscher D, Dekker A: Predicting outcomes in radiation oncology - multifactorial decision support systems. Nat Rev Clin Oncol. 2013, 10: 27-40. 10.1038/nrclinonc.2012.196.
Chin L, Gray JW: Translating insights from the cancer genome into clinical practice. Nature. 2008, 452: 553-563. 10.1038/nature06914.
Khleif SN, Doroshow JH, Hait WN: AACR-FDA-NCI Cancer Biomarkers Collaborative consensus report: advancing the use of biomarkers in cancer drug development. Clin Cancer Res. 2010, 16: 3299-3318. 10.1158/1078-0432.CCR-10-0880.
van’t Veer LJ, Bernards R: Enabling personalized cancer medicine through analysis of gene-expression patterns. Nature. 2008, 452: 564-570. 10.1038/nature06915.
Starmans MH, Pintilie M, John T, Der SD, Shepherd FA, Jurisica I, Lambin P, Tsao MS, Boutros PC: Exploiting the noise: improving biomarkers with ensembles of data analysis methodologies. Genome Med. 2012, 4: 84-10.1186/gm385.
Starmans MH, Fung G, Steck H, Wouters BG, Lambin P: A simple but highly effective approach to evaluate the prognostic performance of gene expression signatures. PLoS One. 2011, 6: e28320-10.1371/journal.pone.0028320.
Venet D, Dumont JE, Detours V: Most random gene expression signatures are significantly associated with breast cancer outcome. PLoS Comput Biol. 2011, 7: e1002240-10.1371/journal.pcbi.1002240.
Boutros PC, Lau SK, Pintilie M, Liu N, Shepherd FA, Der SD, Tsao MS, Penn LZ, Jurisica I: Prognostic gene signatures for non-small-cell lung cancer. Proc Natl Acad Sci U S A. 2009, 106: 2824-2828. 10.1073/pnas.0809444106.
Bentzien J, Muegge I, Hamner B, Thompson DC: Crowd computing: using competitive dynamics to develop and refine highly predictive models. Drug Discov Today. 2013, 18: 472-478. 10.1016/j.drudis.2013.01.002.
Shi L, Campbell G, Jones WD, Campagne F, Wen Z, Walker SJ, Su Z, Chu TM, Goodsaid FM, Pusztai L, Shaughnessy JD, Oberthuer A, Thomas RS, Paules RS, Fielden M, Barlogie B, Chen W, Du P, Fischer M, Furlanello C, Gallas BD, Ge X, Megherbi DB, Symmans WF, Wang MD, Zhang J, Bitter H, Brors B, Bushel PR, Bylesjo M, et al: The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nat Biotechnol. 2010, 28: 827-838. 10.1038/nbt.1665.
Tarca AL, Lauria M, Unger M, Bilal E, Boue S, Kumar Dey K, Hoeng J, Koeppl H, Martin F, Meyer P, Nandy P, Norel R, Peitsch M, Rice JJ, Romero R, Stolovitzky G, Talikka M, Xiang Y, Zechner C: Strengths and limitations of microarray-based phenotype prediction: lessons learned from the IMPROVER Diagnostic Signature Challenge. Bioinformatics. 2013, 29: 2892-2899. 10.1093/bioinformatics/btt492.
Acute Myeloid Leukemia Outcome Prediction Challenge; [https://www.synapse.org/#!Synapse:syn2455683]
Pujana MA, Han JD, Starita LM, Stevens KN, Tewari M, Ahn JS, Rennert G, Moreno V, Kirchhoff T, Gold B, Assmann V, Elshamy WM, Rual JF, Levine D, Rozek LS, Gelman RS, Gunsalus KC, Greenberg RA, Sobhian B, Bertin N, Venkatesan K, Ayivi-Guedehoussou N, Solé X, Hernández P, Lázaro C, Nathanson KL, Weber BL, Cusick ME, Hill DE, Offit K, et al: Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat Genet. 2007, 39: 1338-1349. 10.1038/ng.2007.2.
Taylor IW, Linding R, Warde-Farley D, Liu Y, Pesquita C, Faria D, Bull S, Pawson T, Morris Q, Wrana JL: Dynamic modularity in protein interaction networks predicts breast cancer outcome. Nat Biotechnol. 2009, 27: 199-204. 10.1038/nbt.1522.
Vaske CJ, Benz SC, Sanborn JZ, Earl D, Szeto C, Zhu J, Haussler D, Stuart JM: Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics. 2010, 26: i237-i245. 10.1093/bioinformatics/btq182.
Marbach D, Prill RJ, Schaffter T, Mattiussi C, Floreano D, Stolovitzky G: Revealing strengths and weaknesses of methods for gene network inference. Proc Natl Acad Sci U S A. 2010, 107: 6286-6291. 10.1073/pnas.0913357107.
Marbach D, Costello JC, Kuffner R, Vega NM, Prill RJ, Camacho DM, Allison KR, Consortium D, Kellis M, Collins JJ, Stolovitzky G: Wisdom of crowds for robust gene network inference. Nat Methods. 2012, 9: 796-804. 10.1038/nmeth.2016.
Prill RJ, Saez-Rodriguez J, Alexopoulos LG, Sorger PK, Stolovitzky G: Crowdsourcing network inference: the DREAM predictive signaling network challenge. Sci Signal. 2011, 4: mr7-10.1126/scisignal.2002212.
HPN-DREAM breast cancer network inference challenge; [https://www.synapse.org/#!Synapse:syn1720047]
Maier R, Zimmer R, Kuffner R: A Turing test for artificial expression data. Bioinformatics. 2013, 29: 2603-2609. 10.1093/bioinformatics/btt438.
Canales RD, Luo Y, Willey JC, Austermiller B, Barbacioru CC, Boysen C, Hunkapiller K, Jensen RV, Knight CR, Lee KY, Ma Y, Maqsodi B, Papallo A, Peters EH, Poulter K, Ruppel PL, Samaha RR, Shi L, Yang W, Zhang L, Goodsaid FM: Evaluation of DNA microarray results with quantitative gene expression platforms. Nat Biotechnol. 2006, 24: 1115-1122. 10.1038/nbt1236.
Roberts ND, Kortschak RD, Parker WT, Schreiber AW, Branford S, Scott HS, Glonek G, Adelson DL: A comparative analysis of algorithms for somatic SNV detection in cancer. Bioinformatics. 2013, 29: 2223-2230. 10.1093/bioinformatics/btt375.
Bell AW, Deutsch EW, Au CE, Kearney RE, Beavis R, Sechi S, Nilsson T, Bergeron JJ, Group HTSW: A HUPO test sample study reveals common problems in mass spectrometry-based proteomics. Nat Methods. 2009, 6: 423-430. 10.1038/nmeth.1333.
`t Hoen PA, Friedländer MR, Almlöf J, Sammeth M, Pulyakhina I, Anvar SY, Laros JF, Buermans HP, Karlberg O, Brännvall M, den Dunnen JT, van Ommen GJ, Gut IG, Guigó R, Estivill X, Syvänen AC, Dermitzakis ET, Lappalainen T: Reproducibility of high-throughput mRNA and small RNA sequencing across laboratories. Nat Biotechnol. 2013, 31: 1015-1022. 10.1038/nbt.2702.
Steijger T, Abril JF, Engstrom PG, Kokocinski F, Consortium R, Akerman M, Alioto T, Ambrosini G, Antonarakis SE, Behr J, Bertone P: Assessment of transcript reconstruction methods for RNA-seq. Nat Methods. 2013, 10: 1177-1184. 10.1038/nmeth.2714.
Ransohoff DF: Proteomics research to discover markers: what can we learn from Netflix?. Clin Chem. 2010, 56: 172-176. 10.1373/clinchem.2009.126698.
Waters H: New $10 million X Prize launched for tricorder-style medical device. Nat Med. 2011, 17: 754-
Acknowledgements
We are indebted to the DREAM community for teaching us how to run challenges, and to Sage Bionetworks for their contributions in organizing DREAM challenges. This study was conducted with the support of the Ontario Institute for Cancer Research through funding provided by the Government of Ontario (to PCB). This work was supported by Prostate Cancer Canada and is proudly funded by the Movember Foundation (Grant #RS2014-01). Dr Boutros was supported by a Terry Fox Research Institute New Investigator Award and a CIHR New Investigator Award. This project was supported by Genome Canada through a Large-Scale Applied Project contract. This work was also supported by the Discovery Frontiers: Advancing Big Data Science in Genomics Research program, which is jointly funded by the Natural Sciences and Engineering Research Council (NSERC) of Canada, the Canadian Institutes of Health Research (CIHR), Genome Canada, and the Canada Foundation for Innovation (CFI). NIH grants R01 CA180778 (JMS) and U24-CA143858 (JMS) supported this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
Cite this article
Boutros, P.C., Margolin, A.A., Stuart, J.M. et al. Toward better benchmarking: challenge-based methods assessment in cancer genomics. Genome Biol 15, 462 (2014). https://doi.org/10.1186/s13059-014-0462-7
Published:
DOI: https://doi.org/10.1186/s13059-014-0462-7