
Abstract
T790M mutation is the most common mechanism for resistance to first- and second-generation tyrosine kinase inhibitors (TKI) for epidermal growth factor receptor (EGFR). Several third-generation EGFR mutant selective TKIs are being explored to conquer this resistance. AZD9291 (osimertinib, tagrisso) has been approved for treatment of the metastatic EGFR T790M mutation-positive non-small cell lung cancer. Resistance to AZD9291 has been described. C797S mutation was reported to be a major mechanism for resistance to T790M-targeting EGFR inhibitors. This review summarizes the latest development in identifying the C797S mutation and EAI045, the novel selective inhibitor overcoming the C797S mutant.
Similar content being viewed by others


Background
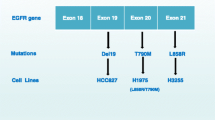
T790M mutation is the most common mechanism of resistance to the first- and second-generation of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKI) [1]. Clinical trials are being done for several T790M-targeting third-generation EGFR-TKIs [2, 3]. These inhibitors include AZD9291 (osimertinib, mereletinib, tagrisso), rociletinib (CO-1686), HM61713 (BI 1482694), ASP8273, EGF816, and PF-06747775 [4–10]. AZD9291 has been shown to have a response rate (RR) of 61 % in EGFR T790M-positive non-small cell lung cancer (NSCLC) patients [4, 10]. HM61713 at 800 mg/day showed a 58.8 % response rate [5]. Unfortunately, these lung cancer patients eventually developed resistance to these drugs after 10 months. A better understanding of the mechanisms of resistance to these third-generation EGFR inhibitors is critical for developing new strategies to treat these patients [11]. EGFR Cys797Ser (C797S) mutation, located within the tyrosine kinase domain, was recently reported to be a potential mechanism of resistance to irreversible EGFR inhibitors such as AZD9291, HM61713, WZ4002, and CO-1686 in T790M-positive patients [12–16] (Fig. 1). This article reviewed the latest development in identifying the C797S mutation and other mechanisms of resistance.

Clonal evolution of NSCLC cancer cells and mechanisms of resistance to third-generation EGFR tyrosine kinase inhibitors. The T790M and C797S mutations were highlighted in the EGFR sequence. Each colored ball represents a distinct clone. The number of balls in each group indicates relative clonal size. NSCLC non-small cell lung cancer, EGFR epidermal growth factor receptor
C797S mediates resistance to AZD9291
In the first-in-human phase I/II AURA trial of AZD9291, systemic progression in NSCLC patients was seen after treatment for a median of 9.6 months [10]. Characterization of the mechanisms of resistance in 22 patients who became resistant to AZD9291 was reported [12]. These patients with progression on AZD9291 in the AURA trial had paired pre-treatment and post-treatment plasma samples. Cell-free DNA (cfDNA) from the plasma of these patients was analyzed by next-gene sequencing (NGS). All EGFR coding exons were analyzed through a 20-gene panel. In the index case, an acquired T → A mutation encoding an EGFR C797S mutation was identified. In another case, an acquired C797S from G → C mutation was documented. This group established a Ba/F3 cell line harboring the C797S mutation and confirmed that the cell line was resistant to AZD9291. Through the study of T790M-positive patients with acquired resistance to AZD9291, three molecular subtypes of AZD9291 resistance were revealed: T790M+/C797S+, T790M+/C797S−, and T790M−/C797S−. The report also discovered that in some cases, two different nucleotide mutations (T to A and G to C) leading to C797S amino acid mutation occurred in the same patients. Since only 6 out of 15 cases acquired C797S mutation, additional mechanisms of resistance to AZD9291 must be present.
In a separate case report, a female non-smoker with widely metastatic lung adenocarcinoma progressed through first-line chemotherapy and second-line erlotinib [17]. She was found to have the EGFR 19 deletion (del 19) and T790M at this point. She was enrolled in the phase 1 AURA study of AZD9291 (NCT01802632) and received AZD9291 for 9 months prior to disease progression. Tumor biopsy at this juncture showed the EGFR C797S mutation, in addition to the del 19 and T790M. Under the strong selective pressure of EGFR-TKIs, the tumor developed secondary T790M and tertiary C797S mutations in the EGFR gene to bypass the TKIs and maintain EGFR signaling.
C797S mutation mediates resistance to HM61713
HM61713 (BI 1482694) is another third-generation EGFR inhibitor and covalently binds to a cysteine residue near the kinase domain of mutant EGFR [18, 19]. In a phase I/II study, HM61713 was shown to be active for patients with T790M-positive NSCLC [5].
The first case report on resistance to HM61713 was on a 57-year-old female never-smoker with stage IV lung adenocarcinoma harboring EGFR del 19 [13]. The patient developed T790M mutation and became refractory to gefitinib. She was enrolled into the trial of HM61713 and was progression free for 17 months. After progression, a repeat biopsy was performed and C797S mutation was found in addition to T790M mutation and del 19. Therefore, the tertiary acquired C797S mutation conferred resistance to another third-generation EGFR TKI.
Exploration of mutations mediating resistance to third-generation TKIs
To search for acquired resistance mutations in EGFR gene, a group from Dana Farber Cancer Center utilized site-directed mutagenesis in EGFR mutant Ba/F3 cell lines harboring sensitizing mutations and/or T790M [14]. The cells were then treated with third-generation TKIs, WZ4002, CO-1686, and AZD9291. Resistant clones were selected out, and mutations were characterized. Three major resistant mutants were identified as EGFR L718Q, L844V, and C797S. All of the three mutations could cause resistance to both WZ4002 and CO-1686. Only C797S mutation confers AZD9291 resistance. Most interestingly, in the presence of del 19 or L858R and T790M, C797S mutation leads to resistance to all current EGFR inhibitors (gefitinib, afatinib, WZ4002, CO-1686, and AZD9291), but L858R/T790M/C797S mutant remains partially sensitive to cetuximab. It remains to be determined whether cetuximab or cetuximab-based combinations are effective clinically in NSCLC patients that develop the L858R/T790M/C797S mutant clone.
In a separate study, a cell line, MGH121, was established from pleural effusion of a NSCLC patient who became resistant to erlotinib [15]. This cell line was sensitive to the third-generation TKIs, including WZ4002, CO-1686, and AZD9291. MGH121 cells were treated with increasing doses of a third-generation TKI, WZ4002. This led to MGH121 Res#1 which was resistant to third-generation TKIs. C797S was found to be the acquired mutation. When the L858R/T790M/C797S mutant construct was stably expressed in MGH121, the cells became resistant to all EGFR TKIs. The study explored further effect of the presence of T790M and C797S together in the same allele (i.e., cis) or in a different allele in the same cell (i.e., trans) on the sensitivity to TKIs. It was clearly demonstrated in the in vitro system that del19/T790M was resistant to the second-generation TKIs, whereas del19/C797S was resistant to the third-generation inhibitors. When T790M and C797S were present in cis, the cells were resistant to all EGFR TKIs. Therefore, characterization of mutation status may guide clinical decision on therapeutic approaches.
HER2 and MET amplification mediates resistance to AZD9291
Since some patients who progressed on AZD9291 were negative for the C797S mutation, additional resistance mechanisms must be present. In a case report, a 54-year-old male with stage IV adenocarcinoma was found to have acquired T790M mutation after progression from second-line treatment with gefitinib [20]. The disease progressed after 12 months of AZD9291 treatment on the AURA trial. HER2 amplification was identified without C797S mutation from the tumor biopsy.
The second patient from the same report was a 60-year-old female, a never-smoker, who was diagnosed with stage IV adenocarcinoma with pleural metastasis [20]. Mutation analysis revealed the known EGFR activating mutation in exon 21, L858R. She received erlotinib and gefitinib sequentially for 12 months. After disease progression, T790M was identified by NGS and she was treated with AZD9291 on the AURA trial [10]. She had partial tumor regression and remained progression free for 10 months. Re-biopsy of the AZD9291 resistant tumor identified an EGFR activating mutation and CMET amplification without T790M or C797S mutation. These two cases indicated that in refractory NSCLC without T790M or C797S mutations, additional gene mutations or amplifications of tyrosine kinases other than EGFR can be the mechanisms of resistance. Additional treatment targeting the mutations will be needed.
EAI045, a fourth-generation selective inhibitor overcoming EGFR C797S
Using purified EGFR mutant kinase peptide containing L858R/T790M mutations, a library of approximately 2.5 million compounds were screened to search for selective inhibitors against the kinase mutant [21]. EGFR allosteric inhibitor-1 (EAI001) was found to have such selectivity toward the EGFR mutant. Further optimization of this compound through medicinal chemistry yielded a highly selective inhibitor, EAI045, toward L858R/T790M mutant (IC50 = 3 nM). EAI045 was found to have a 1000-fold selectivity for the mutant versus wild-type EGFR. The compound is an allosteric inhibitor, rather than an ATP-competing agent. EAI045 was confirmed to be highly selective against a panel of 250 protein kinase peptides. However, EAI045 was not able to completely abolish EGFR autophosphorylation in H1975 NSCLC cell line harboring the L858R/T790M mutant. Since EGFR dimerization is required for kinase enzyme activation [22–24], the investigators hypothesized that EAI045 was active against one subunit of an EGFR heterodimer/asymmetric dimer [21]. It was confirmed that dimerization-defective/independent mutants were markedly more sensitive to EAI045. When combined with cetuximab that blocks EGFR dimerization [25], EAI045 markedly reduced tumor growth in a mouse model of L858R/T790M—mutant-driven lung cancer. The mice treated alone with EAI045 did not respond. EAI045 in combination with cetuximab also induced marked tumor shrinkage in the mouse model carrying L858R/T790M/C797S, a mutant known to be resistant to all third-generation EGFR TKIs. EAI045 and cetuximab exhibited mechanistic synergy. EAI045 represents a novel selective inhibitor that can overcome T790M and C797S resistance mutations [21].
Discussion
C797S mutation in the EGFR gene was found to confer resistance clinically to third-generation TKIs, AZD9291 and HM61713. This likely represents a tertiary acquired mutation that mediates resistance to all known third-generation EGFR TKIs. Some cases were found to harbor two independent clones of C797S mutation, while others even became T790M negative, indicating the heterogeneity of malignant cells. This remains a significant challenge for treatment of lung cancers.
C481S mutation has been reported to mediate resistance to ibrutinib, the first-in-class irreversible Bruton tyrosine kinase (BTK) inhibitor [26–29]. Ibrutinib covalently binds to the cysteine residue 481 in the BTK. This may suggest that the change from cysteine residue to serine may be a recurring mutation that can block inhibitor binding to a broad range of tyrosine kinases. Targeted sequencing for cysteine residue codon mutations may represent a new method to rapidly identify mutations in other tyrosine kinases that harbor similar cysteine-containing motif in the tyrosine kinase domain.
Since more and more TKIs targeting EGFR and anaplastic lymphoma kinase (ALK) mutations are being used for NSCLC therapy [30–34], molecular testing guidance has been established [35]. The guideline suggests that lung cancer tissues are tested by PCR for EGFR mutations and by FISH for ALK mutations [35]. Liquid biopsy is increasingly used for cancer diagnosis and therapy monitoring [36]. cfDNA was used for screening of EGFR mutations. This led to the discovery of C797S mutation [12]. This technology of testing cfDNA is becoming an important companion tool for biomarker analysis and facilitating drug development [37–41].
With the availability of AZD9291 for clinical treatment of T790M-positive NSCLC, more and more resistant cases will appear. Additional mechanisms of resistance to third-generation TKIs, such as HER2 and MET amplification in C797S negative cases, were identified. Loss of T790M mutation was another mechanism of resistance to AZD9291 [12]. Due to the vast diversity of malignant clones, combination therapy of AZD9291 with other agents will be needed to overcome the spectrum of resistant clones [42]. AZD9291 in combination with MET inhibitors or MEK inhibitors is being explored (NCT02143466). Immune checkpoint blockers have been shown to be active in a broad range of malignancies [43–51]. Combination of AZD9291 and PD-L1 antibody is underway in the multi-arm phase Ib study (NCT02143466). Finally, through purposefully targeting allosteric sites in the EGFR tyrosine kinase domain and screening a vast library of compounds that selectively target the resistant EGFR mutant, a highly selective inhibitor, EAI045, has been discovered.
Conclusions
EGFR C797S mutation mediates resistance to third-generation TKIs, AZD9291 and HM61713. Additional mechanisms of resistance are being identified. Combination therapy of AZD9291 with other agents may be one way to overcome the acquired mutation. EAI045 represents a purposefully designed selective inhibitor overcoming EGFR C797S mutation.
Abbreviations
ALK, anaplastic lymphoma kinase; BTK, Bruton tyrosine kinase; cfDNA, cell-free DNA; EGFR, epidermal growth factor receptor; NGS, next-gene sequencing; NSCLC, non-small cell lung cancer
References
Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7(3):169–81.
Chong CR, Janne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 2013;19(11):1389–400.
Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, Cortot AB, Chirieac L, Iacob RE, Padera R, Engen JR, Wong KK, Eck MJ, Gray NS, Janne PA. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462(7276):1070–4.
Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ, Hughes G, Rahi A, Jacobs VN, Red Brewer M, Ichihara E, Sun J, Jin H, Ballard P, Al-Kadhimi K, Rowlinson R, Klinowska T, Richmond GH, Cantarini M, Kim DW, Ranson MR, Pao W. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4(9):1046–61.
Park K, Lee J-S, Lee KH, Kim J-H, Min YJ, Cho JY, Han J-Y, Kim B-S, Kim J-S, Lee DH, Kang JH, Cho EK, Jang I-J, Jung J, Kim H-Y, Sin HJ, Son J, Woo JS, Kim D-W. Updated safety and efficacy results from phase I/II study of HM61713 in patients (pts) with EGFR mutation positive non-small cell lung cancer (NSCLC) who failed previous EGFR-tyrosine kinase inhibitor (TKI). ASCO Meeting Abstracts. 2015;33(15_suppl):8084.
Sequist LV, Rolfe L, Allen AR. Rociletinib in EGFR-mutated non-small-cell lung cancer. N Engl J Med. 2015;373(6):578–9.
Sequist LV, Soria JC, Goldman JW, Wakelee HA, Gadgeel SM, Varga A, Papadimitrakopoulou V, Solomon BJ, Oxnard GR, Dziadziuszko R, Aisner DL, Doebele RC, Galasso C, Garon EB, Heist RS, Logan J, Neal JW, Mendenhall MA, Nichols S, Piotrowska Z, Wozniak AJ, Raponi M, Karlovich CA, Jaw-Tsai S, Isaacson J, Despain D, Matheny SL, Rolfe L, Allen AR, Camidge DR. Rociletinib in EGFR-mutated non-small-cell lung cancer. N Engl J Med. 2015;372(18):1700–9.
Walter AO, Sjin RT, Haringsma HJ, Ohashi K, Sun J, Lee K, Dubrovskiy A, Labenski M, Zhu Z, Wang Z, Sheets M, St Martin T, Karp R, van Kalken D, Chaturvedi P, Niu D, Nacht M, Petter RC, Westlin W, Lin K, Jaw-Tsai S, Raponi M, Van Dyke T, Etter J, Weaver Z, Pao W, Singh J, Simmons AD, Harding TC, Allen A. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 2013;3(12):1404–15.
Wang S, Cang S, Liu D. Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer. J Hematol Oncol. 2016;9(1):34.
Janne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, Ahn MJ, Kim SW, Su WC, Horn L, Haggstrom D, Felip E, Kim JH, Frewer P, Cantarini M, Brown KH, Dickinson PA, Ghiorghiu S, Ranson M. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015;372(18):1689–99.
Costa DB, Kobayashi SS. Whacking a mole-cule: clinical activity and mechanisms of resistance to third generation EGFR inhibitors in EGFR mutated lung cancers with EGFR-T790M. Transl Lung Cancer Res. 2015;4(6):809–15.
Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, Ercan D, Matthews SE, Cantarini M, Barrett JC, Janne PA, Oxnard GR. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015;21(6):560–2.
Song HN, Jung KS, Yoo KH, Cho J, Lee JY, Lim SH, Kim HS, Sun JM, Lee SH, Ahn JS, Park K, Choi YL, Park W, Ahn MJ. Acquired C797S mutation upon treatment with a T790M-specific third-generation EGFR inhibitor (HM61713) in non-small cell lung cancer. J Thorac Oncol. 2016;11(4):e45–7.
Ercan D, Choi HG, Yun CH, Capelletti M, Xie T, Eck MJ, Gray NS, Janne PA. EGFR mutations and resistance to irreversible pyrimidine-based EGFR inhibitors. Clin Cancer Res. 2015;21(17):3913–23.
Niederst MJ, Hu H, Mulvey HE, Lockerman EL, Garcia AR, Piotrowska Z, Sequist LV, Engelman JA. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin Cancer Res. 2015;21(17):3924–33.
Godin-Heymann N, Ulkus L, Brannigan BW, McDermott U, Lamb J, Maheswaran S, Settleman J, Haber DA. The T790M “gatekeeper” mutation in EGFR mediates resistance to low concentrations of an irreversible EGFR inhibitor. Mol Cancer Ther. 2008;7(4):874–9.
Yu HA, Tian SK, Drilon AE, Borsu L, Riely GJ, Arcila ME, Ladanyi M. Acquired resistance of EGFR-mutant lung cancer to a T790M-specific EGFR inhibitor: emergence of a third mutation (C797S) in the EGFR tyrosine kinase domain. JAMA Oncol. 2015;1(7):982–4.
Lee CK, Wu Y-L, Ding PN, Lord SJ, Inoue A, Zhou C, Mitsudomi T, Rosell R, Pavlakis N, Links M, Gebski V, Gralla RJ, Yang JC-H. Impact of specific epidermal growth factor receptor (EGFR) mutations and clinical characteristics on outcomes after treatment with EGFR tyrosine kinase inhibitors versus chemotherapy in EGFR-mutant lung cancer: a meta-analysis. J Clin Oncol. 2015;33(17):1958–65.
Lee K-O, Cha MY, Kim M, Song JY, Lee J-H, Kim YH, Lee Y-M, Suh KH, Son J. Abstract LB-100: discovery of HM61713 as an orally available and mutant EGFR selective inhibitor. Cancer Res. 2014;74(19 Supplement):LB-100.
Planchard D, Loriot Y, Andre F, Gobert A, Auger N, Lacroix L, Soria JC. EGFR-independent mechanisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Ann Oncol. 2015;26(10):2073–8.
Jia Y, Yun CH, Park E, Ercan D, Manuia M, Juarez J, Xu C, Rhee K, Chen T, Zhang H, Palakurthi S, Jang J, Lelais G, DiDonato M, Bursulaya B, Michellys PY, Epple R, Marsilje TH, McNeill M, Lu W, Harris J, Bender S, Wong KK, Janne PA, Eck MJ. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature. 2016;534(7605):129–32.
Red Brewer M, Yun CH, Lai D, Lemmon MA, Eck MJ, Pao W. Mechanism for activation of mutated epidermal growth factor receptors in lung cancer. Proc Natl Acad Sci U S A. 2013;110(38):E3595–604.
Shan Y, Eastwood MP, Zhang X, Kim ET, Arkhipov A, Dror RO, Jumper J, Kuriyan J, Shaw DE. Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization. Cell. 2012;149(4):860–70.
Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, Eck MJ. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007;11(3):217–27.
Cho J, Chen L, Sangji N, Okabe T, Yonesaka K, Francis JM, Flavin RJ, Johnson W, Kwon J, Yu S, Greulich H, Johnson BE, Eck MJ, Janne PA, Wong KK, Meyerson M. Cetuximab response of lung cancer-derived EGF receptor mutants is associated with asymmetric dimerization. Cancer Res. 2013;73(22):6770–9.
Cheng S, Guo A, Lu P, Ma J, Coleman M, Wang YL. Functional characterization of BTK(C481S) mutation that confers ibrutinib resistance: exploration of alternative kinase inhibitors. Leukemia. 2015;29(4):895–900.
Cheng S, Ma J, Guo A, Lu P, Leonard JP, Coleman M, Liu M, Buggy JJ, Furman RR, Wang YL. BTK inhibition targets in vivo CLL proliferation through its effects on B-cell receptor signaling activity. Leukemia. 2014;28(3):649–57.
Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, Grant B, Sharman JP, Coleman M, Wierda WG, Jones JA, Zhao W, Heerema NA, Johnson AJ, Sukbuntherng J, Chang BY, Clow F, Hedrick E, Buggy JJ, James DF, O’Brien S. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32–42.
Novero A, Ravella PM, Chen Y, Dous G, Liu D. Ibrutinib for B cell malignancies. Exp Hematol Oncol. 2014;3(1):1–7.
Iragavarapu C, Mustafa M, Akinleye A, Furqan M, Mittal V, Cang S, Liu D. Novel ALK inhibitors in clinical use and development. J Hematol Oncol. 2015;8(1):17.
Shaw AT, Gandhi L, Gadgeel S, Riely GJ, Cetnar J, West H, Camidge DR, Socinski MA, Chiappori A, Mekhail T, Chao BH, Borghaei H, Gold KA, Zeaiter A, Bordogna W, Balas B, Puig O, Henschel V, study i, Ou SI. Alectinib in ALK-positive, crizotinib-resistant, non-small-cell lung cancer: a single-group, multicentre, phase 2 trial. Lancet Oncol. 2015;17(2):234–42.
Shaw AT, Kim DW, Mehra R, Tan DS, Felip E, Chow LQ, Camidge DR, Vansteenkiste J, Sharma S, De Pas T, Riely GJ, Solomon BJ, Wolf J, Thomas M, Schuler M, Liu G, Santoro A, Lau YY, Goldwasser M, Boral AL, Engelman JA. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370(13):1189–97.
Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F, Wu YL, Thomas M, O’Byrne KJ, Moro-Sibilot D, Camidge DR, Mok T, Hirsh V, Riely GJ, Iyer S, Tassell V, Polli A, Wilner KD, Janne PA. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385–94.
Wu J, Savooji J, Liu D. Second- and third-generation ALK inhibitors for non-small cell lung cancer. J Hematol Oncol. 2016;9(1):19.
Lindeman NI, Cagle PT, Beasley MB, Chitale DA, Dacic S, Giaccone G, Jenkins RB, Kwiatkowski DJ, Saldivar JS, Squire J, Thunnissen E, Ladanyi M. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Thorac Oncol. 2013;8(7):823–59.
Sun W, Yuan X, Tian Y, Wu H, Xu H, Hu G, Wu K. Non-invasive approaches to monitor EGFR-TKI treatment in non-small-cell lung cancer. J Hematol Oncol. 2015;8(1):95.
Smith A, Roda D, Yap T. Strategies for modern biomarker and drug development in oncology. J Hematol Oncol. 2014;7(1):70.
Fernandez-Cuesta L, Perdomo S, Avogbe PH, Leblay N, Delhomme TM, Gaborieau V, Abedi-Ardekani B, Chanudet E, Olivier M, Zaridze D, Mukeria A, Vilensky M, Holcatova I, Polesel J, Simonato L, Canova C, Lagiou P, Brambilla C, Brambilla E, Byrnes G, Scelo G, Le Calvez-Kelm F, Foll M, McKay JD, Brennan P: Identification of circulating tumor DNA for the early detection of small-cell lung cancer. EBio Medicine 2016;8. doi:10.1016/j.ebiom.2016.1006.1032.
Ma M, Shi C, Qian J, Teng J, Zhong H, Han B. Comparison of plasma and tissue samples in epidermal growth factor receptor mutation by ARMS in advanced non-small cell lung cancer. Gene. 2016;591(1):58–64.
Matikas A, Syrigos KN, Agelaki S: Circulating biomarkers in non-small-cell lung cancer: current status and future challenges. Clin Lung Cancer 2016, 17:10.1016/j.cllc.2016.1005.1021.
Hayashi M, Chu D, Meyer CF, Llosa NJ, McCarty G, Morris CD, Levin AS, Wolinsky JP, Albert CM, Steppan DA, Park BH, Loeb DM: Highly personalized detection of minimal Ewing sarcoma disease burden from plasma tumor DNA. Cancer 2016;122. doi:10.1002/cncr.30144.
Niu F-Y, Wu Y-L. Novel agents and strategies for overcoming EGFR TKIs resistance. Exp Hematol Oncol. 2014;3(1):2.
Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, Barlesi F, Kohlhaufl M, Arrieta O, Burgio MA, Fayette J, Lena H, Poddubskaya E, Gerber DE, Gettinger SN, Rudin CM, Rizvi N, Crino L, Blumenschein Jr GR, Antonia SJ, Dorange C, Harbison CT, Graf Finckenstein F, Brahmer JR. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373(17):1627–39.
Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, Waterhouse D, Ready N, Gainor J, Aren Frontera O, Havel L, Steins M, Garassino MC, Aerts JG, Domine M, Paz-Ares L, Reck M, Baudelet C, Harbison CT, Lestini B, Spigel DR. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373(2):123–35.
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–65.
Davar D, Socinski MA, Dacic S, Burns TF. Near complete response after single dose of nivolumab in patient with advanced heavily pre-treated KRAS mutant pulmonary adenocarcinoma. Exp Hematol Oncol. 2015;4(1):34.
Goel G, Sun W. Advances in the management of gastrointestinal cancers—an upcoming role of immune checkpoint blockade. J Hematol Oncol. 2015;8(1):86.
Lin A, Lin E. Programmed death 1 blockade, an Achilles heel for MMR-deficient tumors? J Hematol Oncol. 2015;8(1):124.
Schmid-Bindert G, Jiang T. First-line nivolumab (anti-PD-1) monotherapy in advanced NSCLC: the story of immune checkpoint inhibitors and “the sorcerers apprentice”. Transl Lung Cancer Res. 2015;4(3):215–6.
West H. Nivolumab as first line monotherapy for advanced non-small cell lung cancer: could we replace first line chemotherapy with immunotherapy? Transl Lung Cancer Res. 2014;3(6):400–2.
Tsai K, Daud A. Nivolumab plus ipilimumab in the treatment of advanced melanoma. J Hematol Oncol. 2015;8(1):123.
Acknowledgements
This project was partly supported by the National Natural Science Foundation of China (Grant No. 81101726). SW was a recipient of CAHON Young Investigator Award (www.cahon.org).
Availability of data and materials
The material supporting the conclusion of this review has been included within the article.
Authors’ contributions
DL designed the study. SW and DL drafted the manuscript and finalized the figure. ST and CL assisted in the language editing and revisions. YS provided useful suggestions. All authors read and approved final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
This is not applicable for this review.
Ethics approval and consent to participate
This is not applicable for this review.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, S., Tsui, S.T., Liu, C. et al. EGFR C797S mutation mediates resistance to third-generation inhibitors in T790M-positive non-small cell lung cancer. J Hematol Oncol 9, 59 (2016). https://doi.org/10.1186/s13045-016-0290-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-016-0290-1