
Abstract
Zebrafish is an established model for the study of vertebrate development, and is especially amenable for investigating hematopoiesis, where there is strong conservation of key lineages, genes, and developmental processes with humans. Over recent years, zebrafish has been increasingly utilized as a model for a range of human hematopoietic diseases, including malignancies. This review provides an overview of zebrafish hematopoiesis and describes its application as a model of leukemia and other hematopoietic disorders.
Similar content being viewed by others

Introduction
Zebrafish represents an alternative model for the study of vertebrate development and its perturbation, with powerful molecular, cellular, and genetic approaches available. This model has proven particularly useful for the study of hematopoiesis and its disruption, with zebrafish possessing all major blood cell types, which are generated by similar developmental pathways [1,2]. There is also strong conservation of transcription factors, signaling components, and functional proteins [3-7]. This review overviews zebrafish hematopoiesis and describes the application of this organism to the study of leukemia and other hematopoietic proliferative disorders.
Review
Zebrafish hematopoiesis
Like other vertebrates, zebrafish undergoes distinct waves of hematopoiesis [8]. At 10 h post fertilization (hpf), the ventral lateral mesoderm gives rise to hemangioblasts, bipotential cells expressing both hematopoietic (scl, lmo2, gata2), and vascular (flk1, fli1) transcription factors [9], which become further specified into both hematopoietic and endothelial cells [10].
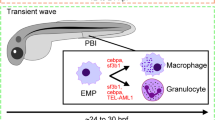
Primitive hematopoiesis is initiated from these cells at two locations, the anterior lateral mesoderm (ALM) and the posterior lateral mesoderm (PLM) that later forms the intermediate cellular mass (ICM) (Figure 1) [11,12]. In the ALM, hemangioblasts differentiate into spi1 + myeloid precursors around 12 hpf, which migrate rostrally and then laterally across the yolk sac [13,14] and begin to express genes encoding cell surface colony-stimulating factor receptors csf1r [15] and csf3r [16]. These mature into distinct myeloid populations with differential expression of the genes for the myeloid-specific actin binding protein l-plastin [15] and the granulocytic enzyme myeloperoxidase (mpo) [17]. In the PLM, gata1 + erythroid precursors are generated at 12 hpf [18]. These commence expression of the erythropoietin receptor (epor) gene and differentiate into primitive erythrocytes expressing erythroid-specific genes including those encoding key enzymes (such as alas2 and carbonic anhydrase) and globins (such as hbbe3) [19], which enter circulation around 24 hpf and persist until 4 dpf [1]. Around 24 hpf, a transient ‘intermediate’ wave of hematopoiesis occurs in the posterior blood island (PBI). Here, bipotent erythromyeloid progenitors (EMP) differentiate into gata1 + erythroid and spi1 + myeloid cells (Figure 1) [13,20].

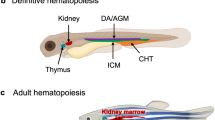
Zebrafish hematopoiesis and its key regulators. Schematic representation of hematopoiesis in zebrafish. The primitive wave commences in two locations, the anterior lateral mesoderm (ALM) (orange), which gives rise to primitive monocytes, and the intermediate cellular mass (ICM) (violet), which generates mostly primitive erythrocytes before 24 hpf. A transient ‘intermediate’ wave occurs in the posterior blood island (PBI) where both erythrocytes and heterophils are formed (grey). Definitive hematopoietic stem cells (HSCs) are initially formed by budding from the hemogenic endothelium on the ventral wall of dorsal aorta (blue). A subset of these HSCs migrate to the caudal hematopoietic tissue (CHT) (yellow) to produce several cell lineages, and also the thymus (purple), where T lymphocyte production occurs. Finally, HSCs seed the developing kidney (green), the final site of definitive hematopoiesis where erythroid, myeloid, and B lymphocyte production occurs. The lineage-specific transcription factors that serve to regulate this process are in red. Abbreviations: BP: B cell progenitor, CLP: common lymphoid progenitor, CMP: common myeloid progenitor, EP: erythroid progenitor, Ery: erythrocyte, GMP: granulocyte-monocyte progenitor, Hemangio: hemangioblast, Hetero: heterophil, HSC: hematopoietic stem cell, Mono: monocyte, TP: T cell progenitor.
Definitive hematopoietic stem cells (HSC) are initially formed at around 33 hpf by budding from the hemogenic endothelium on the ventral wall of the dorsal aorta, equivalent to the aorta-gonad-mesonephros (AGM) in mammals [21,22]. From 48 hpf, a subset of the c-myb + runx1 + HSCs expressing cd44 migrate from the dorsal aorta to the caudal hematopoietic tissue (CHT), equivalent to the fetal liver in mammals [10]. Erythropoiesis commences at 3.5 dpf in the CHT as identified by expression of gata1 and hbbe3, producing mature erythrocytes that gradually replace the primitive erythrocytes in circulation [23]. Definitive myelopoiesis similarly begins around 3 dpf in the CHT, as recognized by l-plastin expression [18].
By 54 hpf, some of the HSCs also migrate directly from the AGM to seed the thymus, the site of lymphopoiesis [24], developing into ikaros + lymphoid progenitors [3]. By 4 dpf cells in the thymus express rag1, a transcription factor essential for lymphoid differentiation [25] and members of the interleukin-2 receptor (IL-2R) family (Sertori et al., submitted), with mature lymphocytes found in the thymus by 7 dpf [26]. Between 48 and 96 hpf, HSCs from the AGM and CHT also seed the marrow of the developing kidney, the ultimate site of definitive hematopoiesis, which is the equivalent of mammalian bone marrow [27,28]. These HSCs produce all hematopoietic lineages, including lymphoid progenitors that mature to B cells in the kidney from 28 dpf [29] and additional T cells in the thymus [1]. The thymus and kidney continue to generate hematopoietic cells throughout adulthood [10].
Zebrafish as a model for leukemia and related disorders
In addition to the strong conservation of key developmental processes and genes, several other factors make zebrafish especially amenable to study the disruptions in hematopoiesis that underpin leukemia and related disorders. Genes can be easily ablated transiently using morpholino-mediated gene knockdown [30] or permanently using genome editing approaches, such as transcription activator-like effector nucleases (TALENs) [31] or clustered regularly interspaced short palindromic repeats (Cas9/CRISPR) [32], or targeting-induced local lesions in genome (TILLING) [33]. Alternatively, genes can be overexpressed transiently by introducing mRNA [34] or DNA [35], or stably through transgenesis, which extends to a variety of conditional/inducible approaches [36]. Sophisticated forward genetic screens are feasible in both zebrafish larvae and adult fish randomly mutated with chemical or transposon-mediated approaches to identify new genes involved in specific biological processes, including disease [37]. The transparent embryos, which develop externally and therefore are extremely accessible, provide exquisite opportunities for imaging [38]. These zebrafish embryo traits are also advantageous for large-scale drug screening by simple immersion as they are penetrable to small molecules and drugs [39,40]. These approaches have yielded new disease models that have increased our knowledge of the oncogenic process and uncovered novel therapeutics (Table 1).
Acute myeloid leukemia models
The first report using zebrafish as a model for leukemogenesis involved enforced expression of the human RUNX1-ETO oncogene, the product of the t(8;21)(q21;q22) translocation found in acute myelogenous leukemia (AML) [41]. Transient overexpression of this fusion gene in zebrafish embryos resulted in blood cell dysplasia, with increased numbers of blast cells, but reduced numbers of c-myb + and hbbe3 + cells, along with circulatory defects [22]. This largely recapitulated the effects seen in mice [42], providing proof-of-principle evidence of the usefulness of this model. This study further showed that knockdown of zebrafish runx1 produced a similar maturation arrest of blood progenitors that accumulated in the ICM, suggesting a likely dominant-negative effect of the RUNX1-ETO fusion protein on endogenous RUNX1 function, providing additional molecular insights [22]. Later, a stable transgenic line was generated that expressed RUNX1-ETO under the control of the zebrafish heat shock-inducible hsp70 promoter. Induction of RUNX1-ETO elicited a fate change within the erythromyeloid lineage toward myelopoiesis, evident by upregulation of spi1 + and mpo + cells at the expense of gata1 + cells. Overexpression of zebrafish scl rescued this phenotype, identifying suppression of SCL as an important mediator of RUNX1-ETO-induced reprogramming, as could treatment with the histone deacetylase inhibitor Trichostatin A [43]. These observations were consistent with both the transcriptional changes and therapeutic sensitivity reported in human AML patients carrying a RUNX1-ETO translocation [41], underscoring the value of zebrafish as a leukemic model. In subsequent chemical suppressor screens performed on this transgenic line, cyclooxygenase inhibitors [44] and the benzodiazepine Ro5-3335 [45] were identified as potential new therapeutics capable of antagonizing the leukemogenic function of RUNX1-ETO (Table 2).
The first bona fide zebrafish model of AML was generated using human MOZ/TIF2, the oncogenic product of inv(8)(p11q13) observed in AML [46]. Transgenic expression of MOZ/TIF2 under the control of the zebrafish white blood cell-specific spi1 promoter led to the development of overt leukemia, characterized by an accumulation of immature myeloid cells in the kidney marrow, but decreased numbers of lymphocyte and precursor cells in the spleen. However, this occurred after a long latency period of 14 to 26 months, with a low incidence of ~1%, suggesting a role for additional genetic mutations [47].
Gain-of-function mutations in the receptor tyrosine kinase FLT3, including internal tandem duplications (FLT3-ITD) and amino acid substitutions in the tyrosine kinase domain (FLT3-TKD), are commonly found in AML [48]. Injection of mRNA encoding patient-derived FLT3-ITD or FLT3-TKD mutants in zebrafish led to constitutive activation of downstream signaling pathways, resulting in expansion of myeloid cells reminiscent of human AML. This was significantly ameliorated by the tyrosine kinase inhibitor AC220 in the case of FLT3-ITD, whereas the effects of FLT3-TKD were resistant to this agent [49].
Mutations in nucleophosmin (NPM1) that result in its aberrant relocation from nucleus to cytoplasm (termed NPMc+) represent one of the most common genetic alterations in adult AMLs [50]. Injection of mRNA encoding human NPMc+, but not NPM1, resulted in perturbation of primitive myelopoiesis, characterized by an increase in spi1 + myeloid precursors with later expansion of mpo + and csf1r + cells, but only in the absence of p53 [51]. This was consistent with the myeloproliferative phenotype reported in a transgenic mouse model expressing the human NPMc+ mutation [52]. Expression of NPMc+ in zebrafish also led to an increase in the number of both erythromyeloid precursors in the PBI and c-myb +/cd41 + HSCs in the ventral wall of the dorsal aorta, suggesting HSCs as the possible cellular origin for NPMc+ leukemia [51].
Members of the MYC family of transcription factors represent some of the most prevalent oncogenes in cancer, including myeloid malignancies, with MYCN overexpression frequently reported in AML patients with a poor prognosis [53]. Transgenic zebrafish expressing the murine Mycn gene under the control of the zebrafish hsp70 promoter developed many aspects of human AML following induction of expression. This included enhanced cell proliferation and accumulation of immature blasts, increased myelopoiesis and anemia, with immature myeloblasts observed in the peripheral blood, kidney marrow, and spleen. Hematopoietic cell fate switch from the erythroid to the myeloid lineage in these transgenic fish was mediated by upregulation of scl and lmo2, leading to decreased expression of gata1 and increased expression of spi1 and mpo, along with ndrg1, a known MYCN target involved in granulocytic maturation [54].
Downregulation of SPI1 expression has been consistently associated with AML [55,56]. Zebrafish carrying a hypomorphic spi1 mutant allele exhibited overproduction of immature granulocytes in the CHT by 3 dpf and in the kidney marrow at later time points, which persisted to 18 months of age. This was associated with a reduction of lymphoid cells in kidney marrow and subsequent accumulation of myeloblasts in the peripheral blood, which closely mimicked aspects of human myelodysplastic syndrome (MDS)/AML. Interestingly, these mutant fish were sensitive to cytarabine, a chemotherapy agent used for the treatment of AML, which further validated the usefulness of zebrafish for therapeutic testing [57].
Finally, zebrafish have been used in a forward genetic screen to identify new genes involved in human hematologic malignancies. This identified a mutation in the ddx18 gene that resulted in a loss of mature myeloid and erythroid cells due to increased apoptosis and p53-dependent G1 arrest. Screening of AML patients identified four non-synonymous sequence variants of DDX18. One of these showed a dominant negative effect over wild-type ddx18 when expressed in zebrafish, validating this approach [58].
Acute lymphoblastic leukemia models
The first stable zebrafish leukemic model was generated by injection of DNA constructs in which sequences encoding mouse c-Myc were fused with those for enhanced green fluorescence protein (EGFP) and placed under the control of the zebrafish lymphoid-specific rag2 promoter. This induced T cell acute lymphoblastic leukemia (T-ALL) with a latency of approximately 7 weeks, with an initial expansion of EGFP+ cells in the thymus, followed by dissemination of these cells into the kidney marrow, spleen, muscle, gut, gills, and fins. Gene expression analysis confirmed that this was the result of clonal expansion of transformed pre-T-lymphoblasts. When these cells were transplanted intraperitoneally into irradiated zebrafish, they homed back to the thymus before infiltrating other organs [2], further validating this as a bona fide leukemic model that has been subsequently utilized for a number of studies. For example, transplantation of the Myc-induced T-ALL cells into a syngenic zebrafish line without immune suppression identified a higher frequency of leukemia-initiating cells in T-ALL than previously thought, suggesting this may also be the case in human T-ALL [34]. This T-ALL syngenic zebrafish model was subsequently used for therapeutic testing, with both cyclophosphamine and vincristine showing efficacy in this model [59]. Collectively, these studies confirmed the similarities between zebrafish and mammalian T-ALL with respect to morphology, genetics and responsiveness to chemotherapic agents.
A conditional zebrafish transgenic line was generated from a similar expression construct, but with a loxed dsRED2 gene inserted upstream of the c-Myc oncogene. Induction of transgene expression by injection of mRNA encoding Cre-recombinase resulted in the development of T-ALL with a disease penetrance of ~13% [60]. Further work in this model demonstrated that enforced expression of zebrafish bcl2 reduced the radiation sensitivity of these T-ALL cells [61]. Using a heat shock-inducible Cre-recombinase, disease penetrance was increased to 81% [62], with a follow-up study showing that co-expression of zebrafish bcl2 in this model accelerated the initiation of T-lymphoblastic lymphoma (T-LBL) but inhibited progression to T-ALL [63], similar to mouse models [64,65]. These effects in zebrafish resulted from elevated levels of sphingosine-1-phosphate receptor 1 and intracellular adhesion molecule 1, which served to increase homotypic cell adhesion and block tumor cell intravasation [63].
An alternate T-ALL model used human MYC fused to a modified estrogen receptor, the nuclear transport of which was controlled by 4-hydroxytamoxifen. Induction of MYC translocation led to T-ALL, with withdrawal of 4-hydroxytamoxifen resulting in complete tumor regression in nearly 75% of fish. This model revealed that MYC suppressed the expression of the tumor suppressor PTEN, resulting in constitutive activation of the AKT pathway to promote tumor progression. Loss-of-function zebrafish pten mutations or expression of a constitutively-active Akt2 rendered tumors MYC-independent [66]. Further studies revealed these effects were mediated by the anti-apoptotic protein BIM [67]. Collectively, these studies both confirmed the importance of survival signals in ALL and suggested AKT pathway inhibitors as a new therapeutic strategy for T-ALL. A subsequent large-scale drug screen using this model identified prephenazine, an anticancer drug used to induce apoptosis in T-ALL cells, as an effective agent. Its effects were shown to be mediated by protein phosphatase 2A, suggesting that pharmacological activation of this enzyme may also have therapeutic potential [68].
The PTEN gene is also frequently mutated in hematological malignancies including T-ALL [69]. Zebrafish pten −/− mutants showed leukemia-like phenotypes, including enhanced proliferation of HSCs within the CHT and differentiation arrest of committed progenitors across the erythroid, myeloid, and lymphoid lineages [70,71]. These phenotypes could be restored by pharmacological inhibition of PI3-K, known to be constitutively activated in the absence of PTEN, using LY294002 [71].
Activating mutations of NOTCH1 have been observed in nearly 60% of T-ALL patients [72]. Overexpression of a T-ALL-derived NOTCH1 intracellular domain mutant under the control of the zebrafish rag2 promoter led to the development of a T cell lymphoproliferative disease. Neoplastic cells invaded tissues and caused a lethal leukemia when transplanted into irradiated recipients. Crossing of these fish with zebrafish overexpressing the bcl2 gene dramatically accelerated the onset of leukemia, indicating a strong cooperation between the NOTCH1 and BCL2 pathways [73].
The ETV6-RUNX1 oncogene, the product of t(1;19)(q23;p13), is frequently observed in B cell ALL [74]. Ubiquitous expression of human ETV6-RUNX1 under the control of the zebrafish β-actin or Xenopus ef-1 promoters resulted in expansion of lymphoid progenitors that resulted in oligoclonal B cell ALL at a frequency of ~3% after a latency period of 8–12 months. These fish showed a dramatic increase in total leukocyte count with more than 90% lymphoblasts observed in blood smears, with enlarged kidneys, the marrow of which were almost completely replaced with lymphoblasts, and dissemination of these cells into the brain, liver, muscle, and ovary, phenotypes reminiscent of childhood pre-B ALL in humans [74]. Lymphoblasts isolated from kidney marrow of these fish caused similar phenotypes in irradiated recipients between 6 and 9 weeks post transplantation, confirming the leukemic nature of these cells. This study identified pro-B precursors as the cellular target of ETV6-RUNX1 fusion, along with downregulation of the ETV6 tumor suppressor gene and deregulation of apoptotic genes, including an altered BCL2:BAX ratio, as potential molecular mediators of ETV6-RUNX1-induced leukemogenesis [75].
Other studies have shown that zebrafish gene orthologues can also recapitulate human oncogenes, further demonstrating conserved functionality across the two species. Alternative ETV6-JAK2 fusions have been identified in both ALL and atypical chronic myelogenous leukemia (CML), that lead to constitutive activation of the JAK2 tyrosine kinase and downstream pathways [76,77]. A fusion was constructed from zebrafish etv6 and jak2a genes corresponding to a human T-ALL-derived ETV6-JAK2. Transient expression of this etv6-jak2a fusion using either the zebrafish spi1 promoter or the ubiquitous cytomegalovirus promoter caused specific disruption of lymphoid cells, with an increased number of rag1 + cells resulting in a fatal lymphoproliferative disorder [78]. The ETV6 gene has also been identified as a tumor-suppressor gene in lymphoid and other malignancies, with its function ablated by both epigenetic and genetic disruption, including the acquisition of truncating mutations [79]. Recent studies have shown that knockdown of zebrafish etv6 resulted in a range of hematopoietic effects, including enhanced lymphopoiesis [80], while transient expression of truncated forms of the etv6 protein exerted similar effects, acting dominantly over the wild-type etv6 protein [81].
To identify other genetic lesions underlying T-ALL, a phenotype-driven forward genetic screen has been performed using chemical mutagenesis of transgenic zebrafish expressing EGFP+ in T lymphocytes. Several mutant lines were identified that recapitulated human T-ALL and T-LBL in both molecular and clinical features [82]. Iterative allo-transplantation of the T-ALL cells, selecting for aggressive disease that models relapsed human T-ALL, yielded leukemic clones that preserved the original genetic aberration in 55% of cases. However, these cells also acquired additional genetic aberrations that might be responsible for their enhanced malignant behavior. More importantly, over 50% of these genes were shared in relapsed human T-ALL samples, reiterating the similar molecular processes governing zebrafish and human hematopoietic malignancies [83].
Myeloproliferative disorder models
Deregulation of the JAK/STAT signaling pathway is associated with a variety of myeloproliferative disorders (MPDs) [84,85]. For example, the somatic activating JAK2 V617F mutation is commonly observed in polycythemia vera and other myeloproliferative disorders [86]. Injection of mRNA encoding the equivalent zebrafish jak2a V581F mutant resulted in enhanced erythropoiesis, with a significant increase in gata1 + precursors, concomitant with increased phosphorylation of the downstream stat5.1 transcription factor. Moreover, the effects of jak2aV581F could be significantly ablated by application of a JAK2 inhibitor, paralleling the clinical situation, as well as knockdown of stat5.1, confirming the importance of this downstream pathway [87]. By corollary, transient expression of a constitutively-active zebrafish stat5.1 H298R / N714F mutant led to the expansion of early and late myeloid cells, erythrocytes, and lymphoid cells [88]. An ETV6-JAK2 fusion gene has also been identified in atypical CML [76]. Expression of the corresponding zebrafish etv6-jak2a fusion gene using either the spi1 or CMV promoter resulted in progenitor hyperproliferation, with increased numbers of immature myeloid and erythroid cells, perturbed mature myeloid cells and anemia, with the lymphoid lineage largely unaffected [89]. This contrasted with the phenotypes observed with the ALL-derived etv6-jak2a fusion, with the functional differences correlating with increased activation of the downstream STAT transcription factors by the atypical CML-derived etv6-jak2a fusion that also showed enhanced sensitivity to JAK2 inhibitors [78].
Activating mutations in RAS genes have been implicated in a number of human proliferative conditions, including the MPD juvenile myelomonocytic leukemia [90]. The first stable model of MPD-utilized heat shock-inducible Cre/Lox-mediated expression of human kRASG12D from the zebrafish β-actin promoter. Whole animal heat shock induction resulted in high tumor incidence with short latency, but associated with significant juvenile lethality and a propensity to develop other disorders, like rhabdomyosarcoma, intestinal hyperplasia, and malignant peripheral nerve sheath tumor. In contrast, ex vivo heat shock of kidney marrow cells specifically elicited a MPD when transplanted into sub-lethally irradiated recipient fish [91]. Expression of the HRASG12V oncogene from the zebrafish fli1 promoter also recapitulated several pathologic aspects of MPD, including defective definitive hematopoiesis, with enhanced proliferation and infiltration of leukocytes into the CHT and kidney marrow. However, no perturbation of the lymphoid lineage was observed, highlighting the lineage-specific function of this oncogene. Expression of HRASG12V was also shown to downregulate NOTCH signaling, which contributed to the proliferation of erythromyeloid progenitors and disease pathogenesis [92].
Activating mutations in the receptor tyrosine kinase encoding c-KIT gene have been observed in systemic mastocytosis (SM), a rare MPD characterized by the infiltration of the clonally derived mast cells into a variety of different tissues including bone marrow [90]. Transgenic expression of the SM-derived KIT D816V mutant under the control of the zebrafish β-actin promoter recapitulated features of aggressive SM, with infiltration of mast cells into the kidney marrow, associated with elevated expression of mast cell proteases, decreased epcam expression, and defects in cell-cycle progression, with significantly increased apoptosis [93].
Myelodysplastic syndrome models
Mutation of the Ten-Eleven Translocation 2 (TET2) gene has been observed in MDS [94]. A tet2 −/− zebrafish line generated through genome editing demonstrated normal embryonic hematopoiesis, but developed a pre-myelodysplasia at 11 months of age. This culminated in true MDS at 24 months, characterized by increased progenitor and myelomonocytic cell numbers, and decreased erythrocyte cell numbers within the kidney marrow [95].
The NUP98-HOXA9 oncogene, the product of t(7;11)(p15;p15), has been observed in MDS, CML blast crisis, and AML [96]. Conditional expression of the human NUP98-HOXA9 oncogene under the control of the zebrafish spi1 promoter was achieved using a Cre/lox-heat shock induction strategy. Heat shock-induced expression of NUP98-HOXA9 perturbed hematopoiesis, with enhanced spi1 + myeloid precursors at the expense of gata1 + erythroid precursors. However, there was also disruption of myeloid differentiation, since mature lcp +, lyz +, and mpo + myeloid cells were not expanded as much as the myeloid precursors. About 23% of the transgenic fish developed myeloproliferative neoplasms between 19 and 23 months, none of which progressed to AML [97].
Recently, the zebrafish crimsonless (crs) mutant was identified in a forward genetic screen that exhibited phenotypes consistent with MDS, including multilineage cytopenia, including decreased hbbe3 + and mpo + cells, disrupted gata1 + precursors, and cell dysplasia. The causal mutation in crs was identified in hspa9b, encoding a conserved mitochondrial matrix chaperone, with the loss-of-function hspa9b G492E mutation disrupting the substrate-binding domain of the protein. This resulted in perturbed mitochondrial function with increased production of reactive oxygen species (ROS) triggering oxidative stress and apoptosis of blood cells, leading to MDS [98].
Conclusions
Zebrafish has emerged as a robust model for human leukemia and related disorders, including AML, ALL, MPD, and MDS. They have proven susceptible to a range of clinically relevant mutations, including those modeled on zebrafish genes, when expressed ubiquitously or in a lineage-specific manner. Alternatively, genetic screening has identified novel genes involved in these diseases. These models have provided unique insights into molecular and cellular aspects of disease etiology, as well as proving effective for therapeutic screening. However, there remains considerable scope for even greater use of this model. The transparency of this organism combined with exquisite transgenic and other fluorescent labeling approaches provides endless opportunities for high resolution in vivo imaging of tumor cell proliferation, dissemination, and interactions with the vasculature and other tissues [99]. The development of highly efficient genome editing approaches expands the potential to interrogate oncogenic and tumor-suppressor pathways and their interactions. In addition, a variety of xenotransplantation approaches are being developed [100], with the generation of immunodeficient and other useful zebrafish strains further enhancing the possibilities. These collectively provide increasingly advanced platforms for pre-clinical therapeutic development, including at the individual patient level, which will underpin advances in individualized medicine.
Abbreviations
- AGM:
-
aorta-gonad-mesonephros
- ALM:
-
anterior lateral mesoderm
- ALL:
-
acute lymphoblastic leukemia
- AML:
-
acute myeloid leukemia
- BP:
-
B cell progenitor
- CHT:
-
caudal hematopoietic tissue
- CLP:
-
common lymphoid progenitor
- CML:
-
chronic myelogenous leukemia
- CMP:
-
common myeloid progenitor
- EGFP:
-
enhance green fluorescent protein
- EMP:
-
erythromyeloid progenitor
- EP:
-
erythroid progenitor
- Ery:
-
erythrocyte
- G-CSFR:
-
granulocyte colony-stimulating factor receptor
- GMP:
-
granulocyte-monocyte progenitor
- Hemangio:
-
hemangioblast
- Hetero:
-
heterophil
- hpf:
-
hours post fertilization
- HSC:
-
hematopoietic stem cell
- ICM:
-
intermediate cell mass
- LBL:
-
lymphoblastic lymphoma
- MDS:
-
myelodysplastic syndrome
- Mono:
-
monocyte/macrophage
- MPD:
-
myeloproliferative disorder
- MPO:
-
myeloperoxidase
- NPM:
-
nucleophosmin
- PBI:
-
posterior blood island
- PLM:
-
posterior lateral mesoderm
- SM:
-
systemic mastocytosis
- TP:
-
T cell progenitor
References
Chen AT, Zon LI. Zebrafish blood stem cells. J Cell Biochem. 2009;108:35–42.
Langenau DM, Traver D, Ferrando AA, Kutok JL, Aster JC, Kanki JP, et al. Myc-induced T cell leukemia in transgenic zebrafish. Science. 2003;299:887–90.
Willett CE, Kawasaki H, Amemiya CT, Lin S, Steiner LA. Ikaros expression as a marker for lymphoid progenitors during zebrafish development. Dev Dyn. 2001;222:694–8.
Oates AC, Pratt SJ, Vail B, Yan Y, Ho RK, Johnson SL, et al. The zebrafish klf gene family. Blood. 2001;98:1792–801.
Lyons SE, Shue BC, Lei L, Oates AC, Zon LI, Liu PP. Molecular cloning, genetic mapping, and expression analysis of four zebrafish c/ebp genes. Gene. 2001;281:43–51.
Liongue C, Ward AC. Evolution of class I cytokine receptors. BMC Evol Biol. 2007;7:120.
Liongue C, Ward AC. Evolution of the Jak-Stat pathway. JAK-STAT. 2013;2:1–8.
Galloway JL, Zon LI. Ontogeny of hematopoiesis: Examining the emergence of hematopoietic cells in the vertebrate embryo. Curr Top Dev Biol. 2003;53:139–58.
Dooley KA, Davidson AJ, Zon LI. Zebrafish scl functions independently in hematopoietic and endothelial development. Dev Biol. 2005;277:522–36.
Paik EJ, Zon LI. Hematopoietic development in the zebrafish. Int J Dev Biol. 2010;54:1127–37.
Davidson AJ, Ernst P, Wang Y, Dekens MP, Kingsley PD, Palis J, et al. cdx4 mutants fail to specify blood progenitors and can be rescued by multiple hox genes. Nature. 2003;425:300–6.
Detrich HW, Kieran MW, Chan FY, Barone LM, Yee K, Rundstadler JA, et al. Intraembryonic hematopoietic cell migration during verterbrate development. Proc Natl Acad Sci U S A. 1995;92:10713–7.
Bennett CM, Kanki JP, Rhodes J, Liu TX, Paw BH, Kieran MW, et al. Myelopoiesis in the zebrafish. Danio rerio Blood. 2001;98:643–51.
Lieschke GJ, Oates AC, Paw BH, Thompson MA, Hall NE, Ward AC, et al. Zebrafish SPI-1(PU.1) marks a site of myeloid development independent of primitive erythropoiesis: implications for axial patterning. Dev Biol. 2002;246:274–95.
Herbomel P, Thisse B, Thisse C. Zebrafish early macrophages colonize cephalic mesenchyme and developing brain, retina, and epidermis through a M-CSF receptor-dependent invasive process. Dev Biol. 2001;238:274–88.
Liongue C, Hall C, O'Connell B, Crozier P, Ward AC. Zebrafish granulocyte colony-stimulating factor receptor signalling promotes myelopoiesis and myeloid cell migration. Blood. 2009;113:2535–46.
Lieschke GJ, Oates AC, Crowhurst MO, Ward AC, Layton JE. Morphologic and functional characterization of granulocytes and macrophages in embryonic and adult zebrafish. Blood. 2001;98:3087–96.
Berman JN, Kanki JP, Look AT. Zebrafish as a model for myelopoiesis during embryogenesis. Exp Hematol. 2005;33:997–1006.
Brownlie A, Hersey C, Oates AC, Paw BH, Falick AM, Witkowska HE, et al. Characterization of embryonic globin genes of the zebrafish. Dev Biol. 2003;255:48–61.
Bertrand JY, Kim AD, Violette EP, Stachura DL, Cisson JL, Traver D. Definitive hematopoiesis initiates through a committed erythromyeloid progenitor in the zebrafish embryo. Development. 2007;134:4147–56.
Burns CE, DeBlasio T, Zhou Y, Zhang J, Zon L, Nimer SD. Isolation and characterization of runxa and runxb, zebrafish members of the runt family of transcriptional regulators. Exp Hematol. 2002;30:1381–9.
Kalev-Zylinska ML, Horsfield JA, Flores MV, Postlethwait JH, Vitas MR, Baas AM, et al. Runx1 is required for zebrafish blood and vessel development and expression of a human RUNX1-CBF2T1 transgene advances a model for studies of leukemogenesis. Development. 2002;129:2015–30.
Jin H, Sood R, Xu J, Zhen F, English MA, Liu PP, et al. Definitive hematopoietic stem/progenitor cells manifest distinct differentiation output in the zebrafish VDA and PBI. Development. 2009;136:647–54.
Le Guyader D, Redd MJ, Colucci-Guyon E, Murayama E, Kissa K, Briolat V, et al. Origins and unconventional behavior of neutrophils in developing zebrafish. Blood. 2008;111:132–41.
Willett CE, Zapata A, Hopkins N, Steiner LA. Expression of zebrafish rag genes during early development identifies the thymus. Dev Biol. 1997;182:331–41.
Trede NS, Zapata A, Zon LI. Fishing for lymphoid genes. Trends Immunol. 2001;22:302–7.
Murayama E, Kissa K, Zapata A, Mordelet E, Briolat V, Lin H-F, et al. Tracing hematopoietic precursor migration to successive hematopoietic organs during zebrafish development. Immunity. 2006;25:963–75.
Bertrand JY, Kim AD, Teng S, Traver D. CD41+ cmyb + precursors colonize the zebrafish pronephros by a novel migration route to initiate adult hematopoiesis. Development. 2008;135:1853–62.
Weinstein JA, Jiang N, White 3rd RA, Fisher DS, Quake SR. High-throughput sequencing of the zebrafish antibody repertoire. Science. 2009;324:807–10.
Bill BR, Petzold AM, Clark KJ, Schimmenti LA, Ekker SC. A primer for morpholino use in zebrafish. Zebrafish. 2009;6:69–77.
Cade L, Reyon D, Hwang WY, Tsai SQ, Patel S, Khayter C, et al. Highly efficient generation of heritable zebrafish gene mutations using homo- and heterodimeric TALENs. Nucleic Acids Res. 2012;40:8001–10.
Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotech. 2013;31:227–9.
Kettleborough RN, Bruijn E, Eeden F, Cuppen E, Stemple DL. High-throughput target-selected gene inactivation in zebrafish. Methods Cell Biol. 2011;104:121–7.
Smith AC, Raimondi AR, Salthouse CD, Ignatius MS, Blackburn JS, Mizgirev IV, et al. High-throughput cell transplantation establishes that tumor-initiating cells are abundant in zebrafish T-cell acute lymphoblastic leukemia. Blood. 2010;115:3296–303.
Hogan BM, Verkade H, Lieschke GJ, Heath JK. Manipulation of gene expression during zebrafish embryonic development using transient approaches. Methods Mol Biol. 2008;469:273–300.
Mosimann C, Zon LI. Advanced zebrafish transgenesis with Tol2 and application for Cre/lox recombination experiments. Methods Cell Biol. 2011;104:173–94.
Patton EE, Zon LI. The art and design of genetic screens: zebrafish. Nature. 2001;2:956–66.
White RM, Sessa A, Burke C, Bowman T, LeBlanc J, Ceol C, et al. Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell. 2008;2:183–9.
Lessman CA. The developing zebrafish (Danio rerio): A vertebrate model for high‐throughput screening of chemical libraries. Birth Defects Res C. 2011;93:268–80.
Peal DS, Peterson RT, Milan D. Small molecule screening in zebrafish. J Cardiovasc Trans Res. 2010;3:454–60.
Lam K, Zhang DE. RUNX1 and RUNX1-ETO: roles in hematopoiesis and leukemogenesis. Front Biosci. 2012;17:1120–39.
Okuda T, Cai Z, Yang S, Lenny N, Lyu CJ, van Deursen JM, et al. Expression of a knocked-in AML1-ETO leukemia gene inhibits the establishment of normal definitive hematopoiesis and directly generates dysplastic hematopoietic progenitors. Blood. 1998;91:3134–43.
Yeh J-RJ, Munson KM, Chao YL, Peterson QP, MacRae CA, Peterson RT. AML1-ETO reprograms hematopoietic cell fate by downregulating scl expression. Development. 2008;135:401–10.
Yeh J-RJ, Munson KM, Elagib KE, Goldfarb AN, Sweetser DA, Peterson RT. Discovering chemical modifiers of oncogene-regulated hematopoietic differentiation. Nat Chem Biol. 2009;5:236–43.
Cunningham L, Finckbeiner S, Hyde RK, Southall N, Marugan J, Yedavalli VR, et al. Identification of benzodiazepine Ro5-3335 as an inhibitor of CBF leukemia through quantitative high throughput screen against RUNX1-CBFbeta interaction. Proc Natl Acad Sci U S A. 2012;109:14592–7.
Deguchi K, Ayton PM, Carapeti M, Kutok JL, Snyder CS, Williams IR, et al. MOZ-TIF2-induced acute myeloid leukemia requires the MOZ nucleosome binding motif and TIF2-mediated recruitment of CBP. Cancer Cell. 2003;3:259–71.
Zhuravleva J, Paggetti J, Martin L, Hammann A, Solary E, Bastie JN, et al. MOZ/TIF2‐induced acute myeloid leukaemia in transgenic fish. Br J Haematol. 2008;143:378–82.
Zeisig BB, Kulasekararaj AG, Mufti GJ, So CW. SnapShot: acute myeloid leukemia. Cancer Cell. 2012;22:698–e1.
He BL, Shi X, Man CH, Ma AC, Ekker SC, Chow HC, et al. Functions of flt3 in zebrafish hematopoiesis and its relevance to human acute myeloid leukemia. Blood. 2014;123:2518–29.
Falini B, Bolli N, Liso A, Martelli M, Mannucci R, Pileri S, et al. Altered nucleophosmin transport in acute myeloid leukaemia with mutated NPM1: molecular basis and clinical implications. Leukemia. 2009;23:1731–43.
Bolli N, Payne EM, Grabher C, Lee J-S, Johnston AB, Falini B, et al. Expression of the cytoplasmic NPM1 mutant (NPMc+) causes the expansion of hematopoietic cells in zebrafish. Blood. 2010;115:3329–40.
Cheng K, Sportoletti P, Ito K, Clohessy JG, Teruya-Feldstein J, Kutok JL, et al. The cytoplasmic NPM mutant induces myeloproliferation in a transgenic mouse model. Blood. 2010;115:3341–5.
Delgado MD, Albajar M, Gomez-Casares MT, Batlle A, Leon J. MYC oncogene in myeloid neoplasias. Clin Transl Oncol. 2013;15:87–94.
Shen L-J, Chen F-Y, Zhang Y, Cao L-F, Kuang Y, Zhong M, et al. Mycn transgenic zebrafish model with the characterization of acute myeloid leukemia and altered hematopoiesis. PLoS One. 2013;8:e59070.
Steidl U, Rosenbauer F, Verhaak RG, Gu X, Ebralidze A, Otu HH, et al. Essential role of Jun family transcription factors in PU. 1 knockdown–induced leukemic stem cells. Nat Genet. 2006;38:1269–77.
Zhu X, Zhang H, Qian M, Zhao X, Yang W, Wang P, et al. The significance of low PU.1 expression in patients with acute promyelocytic leukemia. J Hematol Oncol. 2012;5:22.
Sun J, Liu W, Li L, Chen J, Wu M, Zhang Y, et al. Suppression of Pu.1 function results in expanded myelopoiesis in zebrafish. Leukemia. 2013;27:1913–7.
Payne EM, Bolli N, Rhodes J, Abdel-Wahab OI, Levine R, Hedvat CV, et al. Ddx18 is essential for cell-cycle progression in zebrafish hematopoietic cells and is mutated in human AML. Blood. 2011;118:903–15.
Mizgirev IV, Revskoy S. A new zebrafish model for experimental leukemia therapy. Cancer Biol Ther. 2010;9:895–902.
Langenau DM, Zon LI. The zebrafish: a new model of T-cell and thymic development. Nat Rev Immunol. 2005;5:307–17.
Langenau DM, Jette C, Berghmans S, Palomero T, Kanki JP, Kutok JL, et al. Suppression of apoptosis by bcl-2 overexpression in lymphoid cells of transgenic zebrafish. Blood. 2005;105:3278–85.
Feng H, Langenau DM, Madge JA, Quinkertz A, Gutierrez A, Neuberg DS, et al. Heat‐shock induction of T‐cell lymphoma/leukaemia in conditional Cre/lox‐regulated transgenic zebrafish. Br J Haematol. 2007;138:169–75.
Feng H, Stachura DL, White RM, Gutierrez A, Zhang L, Sanda T, et al. T-lymphoblastic lymphoma cells express high levels of BCL2, S1P1, and ICAM1, leading to a blockade of tumor cell intravasation. Cancer Cell. 2010;18:353–66.
Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331–3.
Kelly PN, Strasser A. The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Diff. 2011;18:1414–24.
Gutierrez A, Grebliunaite R, Feng H, Kozakewich E, Zhu S, Guo F, et al. Pten mediates Myc oncogene dependence in a conditional zebrafish model of T cell acute lymphoblastic leukemia. J Exp Med. 2011;208:1595–603.
Reynolds C, Roderick JE, LaBelle JL, Bird G, Mathieu R, Bodaar K, et al. Repression of BIM mediates survival signaling by MYC and AKT in high-risk T-cell acute lymphoblastic leukemia. Leukemia. 2014;28:1819–27.
Gutierrez A, Pan L, Groen RW, Baleydier F, Kentsis A, Marineau J, et al. Phenothiazines induce PP2A-mediated apoptosis in T cell acute lymphoblastic leukemia. J Clin Inv. 2014;124:644–55.
Gutierrez A, Sanda T, Grebliunaite R, Carracedo A, Salmena L, Ahn Y, et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood. 2009;114:647–50.
Fu CT, Zhu KY, Mi JQ, Liu YF, Murray ST, Fu YF, et al. An evolutionarily conserved PTEN-C/EBPalpha-CTNNA1 axis controls myeloid development and transformation. Blood. 2010;115:4715–24.
Choorapoikayil S, Kers R, Herbomel P, Kissa K, den Hertog J. Pivotal role of Pten in the balance between proliferation and differentiation of hematopoietic stem cells in zebrafish. Blood. 2014;123:184–90.
Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–71.
Chen J, Jette C, Kanki J, Aster J, Look A, Griffin J. NOTCH1-induced T-cell leukemia in transgenic zebrafish. Leukemia. 2007;21:462–71.
Harrison CJ. The detection and significance of chromosomal abnormalities in childhood acute lymphoblastic leukaemia. Blood Rev. 2001;15:49–59.
Sabaawy HE, Azuma M, Embree LJ, Tsai HJ, Starost MF, Hickstein DD. TEL-AML1 transgenic zebrafish model of precursor B cell acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2006;103:15166–71.
Lacronique V, Boureux A, Valle VD, Poirel H, Quang CT, Mauchauffe M, et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science. 1997;278:1309–12.
Lacronique V, Boureux A, Monni R, Dumon S, Mauchauffe M, Mayeux P, et al. Transforming properties of chimeric TEL-JAK proteins in Ba/F3 cells. Blood. 2000;95:2076–83.
Onnebo SM, Rasighaemi P, Kumar J, Liongue C, Ward AC. Alternate TEL-JAK2 fusions associated with T cell acute lymphoblastic leukemia and atypical chronic myelogenous leukemia dissected in zebrafish. Haematologica. 2012;97:1895–903.
Rasighaemi P, Liongue C, Ward AC. ETV6 (TEL1) in blood cell development and malignancy. J Blood Disord. 2014;1:7.
Rasighaemi P, Onnebo SM, Liongue C, Ward AC. ETV6(TEL1) regulates embryonic hematopoiesis in zebrafish. Haematologica. 2015;100:23–31.
Rasighaemi P, Onnebo SMN, Liongue C, Ward AC. Functional analysis of truncated forms of ETV6 (TEL1). Br J Haematol. 2015:(in press) doi:10.1111/bjh.13428.
Frazer JK, Meeker N, Rudner L, Bradley DF, Smith AC, Demarest B, et al. Heritable T-cell malignancy models established in a zebrafish phenotypic screen. Leukemia. 2009;23:1825–35.
Rudner L, Brown K, Dobrinski K, Bradley D, Garcia M, Smith A, et al. Shared acquired genomic changes in zebrafish and human T-ALL. Oncogene. 2011;30:4289–96.
Hayakawa F, Towatari M, Iida H, Wakao H, Kiyoi H, Naoe T, et al. Differential constitutive activation between STAT-related proteins and MAP kinase in primary acute myelogenous leukaemia. Br J Haematol. 1998;101:521–8.
O'Sullivan LA, Liongue C, Lewis RS, Stephenson SEM, Ward AC. Cytokine receptor signaling through the Jak/Stat/Socs pathway in disease. Mol Immunol. 2007;44:2497–506.
Constantinescu SN, Leroy E, Gryshkova V, Pecquet C, Dusa A. Activating Janus kinase pseudokinase domain mutations in myeloproliferative and other blood cancers. Biochem Soc Trans. 2013;41:1048–54.
Ma AC, Fan A, Ward AC, Liongue C, Lewis RS, Cheng SH, et al. A novel zebrafish jak2a(V581F) model shared features of human JAK2(V617F) polycythemia vera. Exp Hematol. 2009;37:1379–86.
Lewis RS, Stephenson SEM, Ward AC. Constitutive activation of zebrafish stat5 expands hematopoietic cell populations in vivo. Exp Hematol. 2006;34:179–87.
Onnebo SMN, Condron MM, McPhee DO, Lieschke GJ, Ward AC. Hematopoietic perturbation in zebrafish expressing a tel-jak2a fusion. Exp Hematol. 2005;33:182–8.
Tefferi A, Gilliland DG. Oncogenes in myeloproliferative disorders. Cell Cycle. 2007;6:550–66.
Le X, Langenau DM, Keefe MD, Kutok JL, Neuberg DS, Zon LI. Heat shock-inducible Cre/Lox approaches to induce diverse types of tumors and hyperplasia in transgenic zebrafish. Proc Natl Acad Sci U S A. 2007;104:9410–5.
Alghisi E, Distel M, Malagola M, Anelli V, Santoriello C, Herwig L, et al. Targeting oncogene expression to endothelial cells induces proliferation of the myelo-erythroid lineage by repressing the notch pathway. Leukemia. 2013;27:2229–41.
Balci TB, Prykhozhij SV, Teh EM, Da'as SI, McBride E, Liwski R, et al. A transgenic zebrafish model expressing KIT-D816V recapitulates features of aggressive systemic mastocytosis. Br J Haematol. 2014;167:48–61.
Ko M, An J, Pastor WA, Koralov SB, Rajewsky K, Rao A. TET proteins and 5-methylcytosine oxidation in hematological cancers. Immunol Rev. 2015;263:6–21.
Gjini E, Mansour MR, Sander JD, Moritz N, Nguyen AT, Kesarsing M, et al. A zebrafish model of myelodysplastic syndrome produced through tet2 genomic editing. Mol Cell Biol. 2014;35:789–804.
Moore MA, Chung KY, Plasilova M, Schuringa JJ, Shieh JH, Zhou P, et al. NUP98 dysregulation in myeloid leukemogenesis. Ann N Y Acad Sci. 2007;1106:114–42.
Forrester AM, Grabher C, McBride ER, Boyd ER, Vigerstad MH, Edgar A, et al. NUP98‐HOXA9‐transgenic zebrafish develop a myeloproliferative neoplasm and provide new insight into mechanisms of myeloid leukaemogenesis. Br J Haematol. 2011;155:167–81.
Craven SE, French D, Ye W, de Sauvage F, Rosenthal A. Loss of Hspa9b in zebrafish recapitulates the ineffective hematopoiesis of the myelodysplastic syndrome. Blood. 2005;105:3528–34.
Moore FE, Langenau DM. Through the looking glass: visualizing leukemia growth, migration, and engraftment using fluorescent transgenic zebrafish. Adv Hematol. 2012;2012:478164.
Bentley VL, Veinotte CJ, Corkery DP, Pinder JB, LeBlanc MA, Bedard K, et al. Focused chemical genomics using zebrafish xenotransplantation as a pre-clinical therapeutic platform for T-cell acute lymphoblastic leukemia. Haematologica. 2015;100:70–6.
Acknowledgments
The authors acknowledge support from the Deakin University International Research Scholarship scheme (PR, FB), the Alfred Deakin Postdoctoral Research Fellow scheme (CL), and the Centre for Molecular and Medical Research (ACW).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
PR and FB drafted the review. CL and ACW conceived the review and contributed to additional input and editing. All authors approved the final version of the manuscript.
Authors’ information
PR and FB are current PhD students and CL is an Alfred Deakin Postdoctoral Research Fellow, all of whom are working on projects related to zebrafish models of leukemia. ACW has a long-standing interest in understanding hematopoiesis and its disruption in a variety of diseases, including malignancy, and has used zebrafish as a model for the majority of these studies.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Rasighaemi, P., Basheer, F., Liongue, C. et al. Zebrafish as a model for leukemia and other hematopoietic disorders. J Hematol Oncol 8, 29 (2015). https://doi.org/10.1186/s13045-015-0126-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-015-0126-4