
Abstract
Background
Partial duplication 1q is a rare cytogenetic anomaly frequently associated to deletion of another chromosome, making it difficult to define the precise contribution of the different specific chromosomal segments to the clinical phenotype.
Case presentation
We report a clinical and cytogenomic study of a patient with multiple congenital anomalies, heart defect, neuromotordevelopment delay, intellectual disability, who presents partial trisomy 1q32 and partial monosomy 11q25 inherited from a paternal balanced translocation identified by chromosome microarray and fluorescence in situ hybridization.
Conclusion
Compared to patients from the literature, the patient’s phenotype is more compatible to the 1q32 duplication’s clinical phenotype, although some clinical features may also be associated to the deleted segment on chromosome 11. This is the smallest 11q terminal deletion ever reported and the first association between 1q32.3 duplication and 11q25 deletion in the literature.
Similar content being viewed by others

Background
Partial duplication 1q is a rare cytogenetic anomaly frequently associated to deletion of another chromosome, making it difficult to define the precise contribution of the different specific chromosomal segments to the clinical phenotype. The duplicated material can result either from an inherited rearrangement or due to an unbalanced de novo translocation, duplication, or insertion [1]. The terminal duplication 1q is associated to pre and postnatal growth retardation, dysmorphisms, multiple congenital malformations, including heart defects, and intellectual disability [2]. Only 12 patients have been described in literature with duplication of the distal long arm of chromosome 1 without other concomitant chromosomal imbalance [3]-[14]. Most cases of these 1q duplication involve the 1q32qter region; and translocations involve multiple chromosomes, including 3p, 5p, 7p, 21q, and Xq [15].
The 11q terminal deletion was first described by Jacobsen et al. [16] and has a recognized pattern of malformation. Jacobsen syndrome (JBS, OMIM 147791) is a contiguous gene disorder caused by 7 to 20 Mb 11qter deletions. More than 100 cases have been described in literature and the clinical findings include developmental delay, short stature, congenital heart defects (CHD), thrombocytopenia, genitourinary anomalies, pyloric stenosis, and ophthalmologic anomalies.
Here we report on a patient with a duplication of 37 Mb of the distal long arm of chromosome1 from 1q32 to 1qter associated to a 2 Mb 11q25qter deletion inherited from his father’s balanced translocation. SNP-array and fluorescence in situ hybridization (FISH) techniques were used to define the specific breakpoints. The patient’s phenotype was reviewed and compared with previous patients with distal 1q duplication and distal 11q deletion.
Case presentation
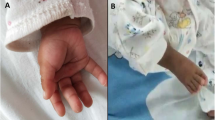
The Research Ethics Committee of UNIFESP approved this study, and consent was obtained from the patient’s parents. The male patient is the first child of a young non-consanguineous and healthy couple and has one normal sister and one paternal half-sister. He was born at term by vaginal delivery after a pregnancy with hypertension and a bleeding episode, with a birth weight of 1,980 g (10th centile), length 42 cm (10th-25th centile), and occipital frontal circumference (OFC) unavailable. He stayed 11 days in the nursery because of low birth weight and dysmorphisms. Hematologic abnormalities were not observed during the neonatal period. At the age of three months, a computerized cranial tomography showed hydrocephaly and at age of six months, the echocardiography showed pulmonary stenosis. At one year old, he had endocarditis, being hospitalized for 18 days. He also had three episodes of bronchopneumonia by the age of two years. At one year and 10 months old, he presented height of 72 cm (<5th centile), weight of 7,200 g (<5th centile), OFC of 47 cm (10th-25th centile). The clinical evaluation revealed: microsomia, relative macrocephaly, brachycephaly, downslanted palpebral fissures, long and marked eyelashes, synophrys, large ears, hipoplastic ala nasi, prominent nose, high nasal bridge, long and smooth philtrum, abnormal tooth implantation, high palate, forehead abnormal hair implantation, hirsutism, pectus carinatum, thoracic asymmetry, vertical palmar creases one on the left hand and two on the right, overlying position of toes, micropenis and bilateral cryptorchidism (Figure 1a). He evolved with moderate development and speech delay, and had a few convulsion episodes at age of two years. A new cardiac evaluation showed atrial septal defect, which was surgically corrected at age of 5 years. The clinical evaluation at age of ten years showed: height 119 cm (<5th centile), weight 25 kg (5th centile) and OFC 50.5 cm (50th centile). At this time, the patient additionally presented muscular hypotonia and presented with moderate intellectual disability, aggressive and hyperactive behavior, limited verbal language repertoire and dysarthria. At age of 14 years, he showed height 125 cm (<5th centile), weight 26 kg (5th centile) and OFC 52.5 cm (50th centile) (Figure 1b). At this time, the patient underwent an assessment of intellectual and neuropsychological skills and had a very limited verbal repertoire, but showed communicative intention, good eye contact and used some gestures to express his intentions. Other behavioral characteristics included mild psychomotor disabilities and lack of symbolic skills, indicating severe cognitive impairment. In daily life activities, according to the mother, he needs supervision.

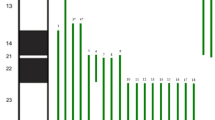
Patient at 1 year and 10 months (a) and 14 years of age (b) showing the facial dysmorphic features; Partial G-banding karyotype showing paternal balanced translocation (c); Partial FISH metaphase with WCP1 probe showing the patient’s der(11) with the duplicated segment (d); BAC-FISH results showing the breakpoint delineation in 1q32.3 and 11q25 regions in father’s metaphases (e and f); Array result for duplication (blue bar) 1q32.(212,508,954-249,224,376) × 3 (g); and deletion (red bar) 11q25(132,927,027-134,944,770) × 1 (h).
Chromosome analysis from lymphocyte cultures revealed additional material in chromosome 11q. The patient’s mother had a normal karyotype but the father carries a balanced translocation t(1;11)(q32;q25) (Figure 1c). Breakpoints were defined by FISH with bacterial artificial chromosome probes (BACs). FISH analyses were performed with whole chromosome paint (WCP1-Cytocell, Cambridge, UK) and using different BAC probes from chromosomes 1 and 11, as previously reported (Figure 1d, e and f) [17]. Further investigations using Genome-Wide Human SNP 6.0 array (Affymetrix Inc., Santa Clara, CA, USA) on the patient revealed the final cytogenomic result as:
46,XY.ish der(11)t(1;11)(q32.3;q25)pat(wcp1+,RP11-663C5-,RP11-262H5+,RP11-15 J5+,RP11-265 F9-). arr[hg19]1q32.3q44(212,508,954-249,224,376) × 3,11q25 (132,927,027-134,944,770) × 1 (Figure 1g and h).
Conclusions
This is the first report of a patient with concomitant 1q32 duplication and 11q25 deletion in the literature. The clinical features observed in our patient are more closely similar to 1q32 duplication than to 11q25 deletion. Comparing our patient with the 13 patients from literature observed by Balasubramanian et al. [14] we concluded that he presents important clinical features observed in the “Distal Trisomy 1q Syndrome”, such as marked developmental delay, microsomia, prominent forehead, downslanted palpebral fissures, long lashes, relative macrocephaly, recurrent pulmonary infections, genitourinary anomalies and CHD. However, the wide range and variety of the clinical features observed in patients with 1q32 duplication make the genotype-phenotype correlation extremely difficult.
The 1q32 duplicated segment in our patient presents 467 genes, including: TGFB2 gene (transforming growth factor, beta 2), RYR2 gene (ryanodine receptor 2), and TBCE gene (Homo sapiens tubulin folding cofactor E).The TGFB2 gene regulates proliferation, differentiation, adhesion, migration, and other functions in many cell types and disruption of the TGFB/SMAD pathway, regulated by this gene, has been implicated in a variety of human cancers. The knockout of this gene in results in perinatal mortality and a wide range of developmental malformations, including CHD. The TGFB2 gene is also involved with Loyes-Dietz syndrome 4 (OMIM # 614816) caused by heterozygous mutation in the TGFB2 gene. Lindsay et al. [18] identified two unrelated patients with aortic aneurysm and de novo 1q41 microdeletions encompassing the TGFB2, one with Marfan syndrome and the other with Loyes-Dietz syndrome-like features. The RYR2 gene, which maps to 1q42q43, encodes a ryanodine receptor found in cardiac muscle. Mutations in this gene are associated with stress-induced polymorphic ventricular tachycardia and arrhythmogenic right ventricular dysplasia. Priori et al. [19] identified RYR2 gene mutations in 12 patients with typical catecholaminergic polymorphic ventricular tachycardia and speculated it as a likely candidate for this genetically transmitted arrhythmic disorder. This gene is not involved in conotruncal congenital malformations as observed in our patient and other patients with 1q32 duplication. The genes RYR2 and VTSIP are also associated to CHD. Other genes labeled in the 1q32.3qter duplicated segment also play an important role in neurodevelopment such as TBCE gene located in 1q43.2.
The 11q terminal deletion disorder (Jacobsen syndrome) presents a recognized pattern of malformation, which depends on the size of the 11qter deletion, usually ranging from 7 to 20 Mb. Since the first report in 1973 by Jacobsen et al. [16] more than 100 cases have been reported in literature. The main clinical features include motor developmental delay, pre and postnatal growth retardation, trigonocephaly, thrombocyto- or pancytopenia, cardiac defects, hypertelorism, downslanted palpebral fissures, short nose, anteverted nares, thin upper lip, V-shaped mouth, dental anomalies, and low-set malformed ears [20]. Evers et al. [21] described a 2-year-old boy with a 2.2 Mb deletion of 11q25qter and a 15.7 Mb terminal duplication of 16q22.3qter, who has no heart disease or platelet dysfunction, which are typically found in more than 90% of patients with terminal 11q deletion. This suggests that the deletion presented in their patient does not encompass the critical region for the Jacobsen syndrome. Our patient presents a smaller 2 Mb deletion than Evers et al. [21] patient, but shares some main clinical features of Jacobsen syndrome, such as cardiac congenital malformation, motor developmental delay, short stature, undescended testes, high forehead, ocular hypertelorism, downslanted palpebral fissures, anteverted nares, thick eyebrows and high-arched palate, but he has no hematologic abnormalities as observed in patients with Jacobsen syndrome. However, some of these clinical features are also described in duplication 1q32 making it extremely difficult to determine the accurate genotype-phenotype correlation. The 11q deletion observed in our patient is the smallest segment described in literature and encompasses 25 genes including the JAM3 (junctional adhesion molecule 3) gene, a candidate gene for the Jacobsen syndrome cardiac phenotype [22] and conotruncal heart defects. This gene may be related to the atrial septal defect presented by our patient.
An early conclusion from the genotype–phenotype findings was that CHD are strongly associated with an altered dose of genes that act together. And most important, the new cytogenomic approaches enabled the detailed study of single cases and provided additional information to the genotype-phenotype correlation.
Consent
Written informed consent was obtained from the parents of the patient for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Abbreviations
- CHD:
-
Congenital heart defects
- JAM3 :
-
Junctional adhesion molecule 3 gene
- OFC:
-
Occipital frontal circumference
- RYR2 :
-
Ryanodine receptor 2 gene
- TBCE :
-
Homo sapiens tubulin folding cofactor E
- TGFB2 :
-
Tubulin folding cofactor E gene
- VTSIP :
-
Ventricular tachycardia, stress-induced polymorphic gene
References
Emberger W, Petek E, Kroisel PM, Zierler H, Wagner K: Clinical and molecular cytogenetic characterization of two patients with partial trisomy 1q41-qter: further delineation of partial trisomy 1q syndrome. Am J Med Genet 2001, 104: 312–318. 10.1002/ajmg.10096
Schinzel A: Catalogue of Unbalanced Chromosome Aberrations in Humans. Edited by Walter de Gruyter, Berlin; 2001.
Steffensen DM, Chu EH, Speert DP, Wall PM, Meilinger K, Kelch RP: Partial trisomy of the long arm of human chromosome 1 as demonstrated by in situ hybridization with 5S ribosomal RNA. Hum Genet 1977, 36: 25–33. 10.1007/BF00390432
Flatz S, Fonatsch C: Partial trisomy 1q due to tandem duplication. Clin Genet 1979, 15: 541–542. 10.1111/j.1399-0004.1979.tb00839.x
Lungarotti MS, Falorni A, Calabro A, Passalacqua F, Dallapiccola B: De novo duplication 1q32-q42: variability of phenotypic features in partial 1q trisomics. J Med Genet 1980, 17: 398–402. 10.1136/jmg.17.5.398
Rasmussen SA, Frias JL, Lafer CZ, Eunpu DL, Zackai EH: Partial duplication 1q: report of four patients and review of the literature. Am J Med Genet 1990, 36: 137–143. 10.1002/ajmg.1320360203
Clark BJ, Lowther GW, Lee WR: Congenital ocular defects associated with an abnormality of the human chromosome 1: trisomy 1q32-qter. J Pediatr Ophthalmol Strabismus 1994, 31: 41–45.
Duba HC, Erdel M, Löffler J, Bereuther L, Fischer H, Utermann B, Utermann G: Detection of a de novo duplication of 1q32-qter by fluorescence in situ hybridisation in a boy with multiple malformations: further delineation of the trisomy 1q syndrome. J Med Genet 1997, 34: 309–313. 10.1136/jmg.34.4.309
De Brasi D, Rossi E, Giglio S, D’Agostino A, Titomanlio L, Farina V, Andria G, Sebastio G: Inv dup del (1)(pter > q44::q44 > q42:) with the classical phenotype of trisomy 1q42-qter. Am J Med Genet 2001, 104: 127–130. 10.1002/ajmg.1589
Nowaczyk MJ, Bayani J, Freeman V, Watts J, Squire J, Xu J: De novo 1q32q44 duplication and distal 1q trisomy syndrome. Am J Med Genet 2003, 120: 229–233. 10.1002/ajmg.a.20028
Polityko A, Starke H, Rumyantseva N, Claussen U, Liehr T, Raskin S: Three cases with rare interstitial rearrangements of chromosome 1 characterized by multicolor banding. Cytogenet Genome Res 2005, 111: 171–174. 10.1159/000086388
Coccé MC, Villa O, Obregon MG, Salido M, Barreiro C, Solé F, Gallego MS: Duplication dup(1)(q41q44) defined by fluorescence in situ hybridization: delineation of the ‘trisomy 1q42 > qter syndrome’. Cytogenet Genome Res 2007, 118: 84–86. 10.1159/000106446
Kulikowski LD, Bellucco FT, Nogueira SI, Christofolini DM, Smith MA, de Mello CB, Brunoni D, Melaragno MI: Pure duplication 1q41-qter: further delineation of trisomy 1q syndromes. Am J Med Genet 2008, 146: 2663–2667. 10.1002/ajmg.a.32510
Balasubramanian M, Barber JC, Collinson MN, Huang S, Maloney VK, Bunyan D, Foulds N: Inverted duplication of 1q32.1 to 1q44 characterized by array CGH and review of distal 1q partial trisomy. Am J Med Genet 2009, 149: 793–797. 10.1002/ajmg.a.32463
Utine GE, Aktas D, Alanay Y, Gücer S, Tuncbilek E, Mrasek K, Liehr T: Distal partial trisomy 1q: report of two cases and a review of the literature. Prenat Diagn 2007, 27: 865–871. 10.1002/pd.1788
Jacobsen P, Hauge M, Henningsen K, Hobolth N, Mikkelsen M, Philip J: An (11;21) translocation in four generations with chromosome 11 abnormalities in the offspring. A clinical, cytogenetical, and gene marker study. Hum Hered 1973, 23: 568–585. 10.1159/000152624
Kulikowski LD, Christ LA, Nogueira SI, Brunoni D, Schwartz S, Melaragno MI: Breakpoint mapping in a case of mosaicism with partial monosomy 9p23 > pter and partial trisomy 1q41 > qter suggests neo-telomere formation in stabilizing the deleted chromosome. Am J Med Genet 2006, 1: 82–87. 10.1002/ajmg.a.31045
Lindsay ME, Schepers D, Bolar NA, Doyle J, Gallo E, Fert-Bober J, Kempers MJE, Fishman EK, Chen Y, Myers L, Bjeda D, Oswald G, Elias AF, Levy HP, Anderlid BM, Yang MH, Bongers EMHF, Timmermans J, Braverman AC, Canham N, Mortier GR, Brunner HG, Byers PH, Van Eyk J, Van Laer L, Dietz HC, Loeys BL: Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nature Genet 2012, 44: 922–927. 10.1038/ng.2349
Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V, Danieli GA: Mutations in the cardiac ryanodine receptor gene ( hRyR2 ) Underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 2001, 103: 196–200. 10.1161/01.CIR.103.2.196
Grossfeld PD, Mattina T, Lai Z, Favier R, Jones KL, Cotter F, Jones C: The 11q terminal deletion disorder: a prospective study of 110 cases. Am J Med Genet 2004, 129: 51–61. 10.1002/ajmg.a.30090
Evers C, Janssen JW, Jauch A, Bonin M, Moog U: A small terminal deletion 11q in a boy without Jacobsen syndrome: narrowing the critical region for the 11q Jacobsen syndrome phenotype. Am J Med Genet 2012, 158: 680–684. 10.1002/ajmg.a.34433
Phillips HM, Renforth GL, Spalluto C, Hearn T, Curtis AR, Craven L, Havarani B, Clement-Jones M, English C, Stumper O, Salmon T, Hutchinson S, Jackson MS, Wilson DI: Narrowing the critical region within 11q24-qter for hypoplastic left heart and identification of a candidate gene, JAM3, expressed during cardiogenesis. Genomics 2002, 79: 475–478. 10.1006/geno.2002.6742
Acknowledgement
This work was supported by grants from Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP): Grants number 2010/05123-5 and 2009/53105-9.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MIM and LDK coordinated and participated in the design of the study. VAM, ALP and CBM carried out the clinical assessments. SST carried out chromosomal classic and FISH analysis and LDK carried out microarray analysis. VAM drafted the manuscript and all authors read and approved the final manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
Cite this article
Meloni, V.A., Takeno, S.S., Pilla, A.L. et al. Trisomy 1q32 and monosomy 11q25 associated with congenital heart defect: cytogenomic delineation and patient fourteen years follow-up. Mol Cytogenet 7, 57 (2014). https://doi.org/10.1186/s13039-014-0057-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-014-0057-8