
Abstract
Background
Cardiac hypertrophy is characterized by alterations in both cardiac bioenergetics and insulin sensitivity. Insulin promotes glucose uptake by cardiomyocytes and its use as a substrate for glycolysis and mitochondrial oxidation in order to maintain the high cardiac energy demands. Insulin stimulates Ca2+ release from the endoplasmic reticulum, however, how this translates to changes in mitochondrial metabolism in either healthy or hypertrophic cardiomyocytes is not fully understood.
Results
In the present study we investigated insulin-dependent mitochondrial Ca2+ signaling in normal and norepinephrine or insulin like growth factor-1-induced hypertrophic cardiomyocytes. Using mitochondrion-selective Ca2+-fluorescent probes we showed that insulin increases mitochondrial Ca2+ levels. This signal was inhibited by the pharmacological blockade of either the inositol 1,4,5-triphosphate receptor or the mitochondrial Ca2+ uniporter, as well as by siRNA-dependent mitochondrial Ca2+ uniporter knockdown. Norepinephrine-stimulated cardiomyocytes showed a significant decrease in endoplasmic reticulum-mitochondrial contacts compared to either control or insulin like growth factor-1-stimulated cells. This resulted in a reduction in mitochondrial Ca2+ uptake, Akt activation, glucose uptake and oxygen consumption in response to insulin. Blocking mitochondrial Ca2+ uptake was sufficient to mimic the effect of norepinephrine-induced cardiomyocyte hypertrophy on insulin signaling.
Conclusions
Mitochondrial Ca2+ uptake is a key event in insulin signaling and metabolism in cardiomyocytes.
Similar content being viewed by others

Background
Cardiac hypertrophy is a physiological process that occurs in response to an increase in heart workload. Initially, it can be compensatory, however, a chronic elevated workload, the neurohumoral input, or an underlying genetic mutation can lead to pathological hypertrophy, contractile dysfunction, ventricular dilatation, and finally, heart failure [1]. Moreover, during pathological hypertrophy, the heart undergoes significant changes in metabolism [2]. Cardiac metabolism is flexible, with fuel preferences switching from primarily glycolysis in the foetal heart, to β-oxidation of lipids in the adult heart [3]. During pathological hypertrophy, there is a re-activation of the so called "foetal gene program" and an increased preference for glucose oxidation [4],[5].
The cellular signalling pathways involved in the hypertrophic response have been widely studied and Ca2+ is known to play a key role [1]. Diverse proteins activated by an elevation in cytosolic Ca2+ have been associated with the development of cardiac hypertrophy, such as the protein phosphatase calcineurin, Ca2+-calmodulin kinase II and protein kinase C [6]-[8]. Mitochondria are another important target of Ca2+ signalling. Mitochondrial Ca2+ uptake is essential for cell bioenergetics; thus an equilibrated rise in mitochondrial Ca2+ concentration stimulates Krebs cycle activity, increasing NADH levels and ATP synthesis [9],[10]. Mitochondrial Ca2+ uptake is carried out primarily by the mitochondrial Ca2+ uniporter (MCU) [11]. This uniporter is a highly selective Ca2+ channel, but with low affinity [12]. Thereby, high cytosolic-Ca2+ concentrations in the proximity of mitochondria must occur to induce Ca2+ entry. To solve this problem, mitochondria are strategically localized to high Ca2+ concentration microdomains near Ca2+ release channels of the endoplasmic reticulum (ER) [13],[14]. The close contacts formed between ER and mitochondria are regulated by a great variety of proteins located in the interface of the two organelles, including the inositol 1,4,5-triphosphate receptor (InsP3R), the ryanodine receptor (RyR), the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) and mitofusin-2 (Mfn2) [15],[16].
Insulin is a key regulator of metabolism and plasma glucose levels [17]. In the heart, insulin promotes glucose uptake into cardiomyocytes and its use as an energy source by activating glycolysis and mitochondrial oxidative phosphorylation [18]. Recently, we showed that insulin increases cytoplasmic Ca2+ levels by a mechanism that involves the sequenced activation of phosphatidilinositol-3 kinase (PI3K) and phospholipase C (PLC) to increase the mass of InsP3. This axis induces Ca2+ release from the InsP3R, that can influence translocation of the facilitated glucose transporter GLUT4 to cell surface and glucose uptake [19]. Mitochondrial Ca2+ uptake can also affect cytoplasmic Ca2+ signals, by acting as a Ca2+ buffer after an increase in cytoplasmic levels. Moreover, it has been shown that, in addition to helping buffer large cytoplasmic Ca2+ fluctuations, the maintenance of optimal mitochondrial function is dependent on continual, constitutive Ca2+ transfer from ER to mitochondria to support oxidative phosphorylation [9]. During pathological cardiac hypertrophy, the heart develops insulin resistance, resulting in a slower rate of glucose entry in response to insulin and a decrease in the capacity for glucose oxidation [20],[21], without changes in the expression of GLUT4 transporters [22].
The current study was designed to understand the mechanisms integrating mitochondrial Ca2+ uptake and insulin signaling in normal and hypertrophic cardiomyocytes. We show that insulin-induced stimulation of oxidative metabolism occurs through mitochondrial uptake of the Ca2+ released from ER. This mechanism is diminished in norepinephrine (NE)-treated cardiomyocytes undergoing pathological hypertrophy, but not in insulin like growth factor-1 (IGF-1)-treated hypertrophic cardiomyocytes. Moreover, pharmacological inhibition of mitochondrial Ca2+ uptake reduces insulin-dependent activation of the canonical Akt pathway. Thus, Ca2+ transfer to mitochondria emerges as an important new regulator of insulin signaling.
Results
Insulin induces an increase in mitochondrial Ca2+ uptake
Previous reports showed that insulin stimulation leads to an increase in cytoplasmic Ca2+ levels by releasing Ca2+ from the ER [19]. However, it is unknown whether the Ca2+ signal triggered by insulin has a mitochondrial component. To examine this, changes in mitochondrial Ca2+ levels were measured in cardiomyocytes using the mitochondrial-directed Ca2+ indicator Rhod-FF. This probe had a Pearson coefficient of 0.92 with the mitochondrial probe MitoTracker Green in colocalization analysis, showing a high degree of mitochondrial destination (Additional file 1: Figure S1A). Once loaded with Rhod-FF, changes in mitochondrial Ca2+ levels were measured via confocal microscopy. As a control, at the end of each measurement, carbonyl cyanide m-chlorophenyl hydrazone (CCCP) was added to dissipate mitochondrial membrane potential (ΔΨM) releasing mitochondria-localized Ca2+ to the cytoplasm (Additional file 1: Figure S1B). Because Rhod-FF at high concentrations can alter mitochondrial morphology [23] we also analyzed mitochondrial mean volume and number [24],[25] in cells treated with MitoTracker Green and Rhod-FF (Additional file 1: Figure S1C-D). No significant differences were found in these parameters suggesting that under our conditions, treatment with Rod-FF did not impact mitochondrial morphology.
As shown in Figure 1A, mitochondrial Ca2+ levels increased in cardiomyocytes after insulin stimulation. Pharmacological inhibition was used to test the participation of various Ca2+ channels. It has been shown that MCU is one of the primary Ca2+ channels involved in mitochondrial Ca2+ uptake in cardiomyocytes [26]. To test whether MCU participates in insulin-induced mitochondrial Ca2+ uptake, cardiomyocytes were pre-treated with the MCU inhibitor Ruthenium Red (Ru) for 30 min prior insulin stimulation. As shown in Figure 1B, there was a significant reduction in mitochondrial Ca2+ uptake after insulin stimulation in Ru-treated cardiomyocytes. Next, participation of InsP3R was evaluated by pre-treating cardiomyocytes with an InsP3R–specific inhibitor xestospongin C (XeC). XeC also reduced the insulin-induced mitochondrial Ca2+ signal (Figure 1C). To further confirm the participation of InsP3R, PLC was blocked with U73122, thus inhibiting InsP3R Ca2+ release by preventing the synthesis of its ligand. Figure 1D shows that cardiomyocytes pre-treated with U73122 had a reduced mitochondrial Ca2+ signal after insulin stimulation. To evaluate whether RyR hannels also contribute to this mitochondrial Ca2+ signal, RyR channels were inhibited by preincubating cardiomyocytes with ryanodine. Figure 1E shows that the insulin-induced increase in mitochondrial Ca2+ was unaltered in cardiomyocytes pre-treated with ryanodine. In conclusion, Ru, XeC and U73122 pre-treatment caused a significant reduction in the area under the curve of the insulin-induced mitochondrial Ca2+ signal, whereas ryanodine did not (Figure 1F).

Insulin increases mitochondrial Ca2+through an InsP 3 R dependent pathway. A) Confocal images of insulin-induced mitochondrial Ca2+ signal captured at the indicated times in cardiomyocytes loaded with Rhod-FF 5,4 μM. B-E) Representative measures of fluorescence-associated insulin-induced mitochondrial Ca2+ signals in cardiomyocytes pre-treated with (gray line) or without (Ctrl, black line) the respective inhibitors for 30 min. Insulin (100 nM) was added at 100 s. B) Ruthenium Red (Ru), 10 μM; C) xestospongin C (XeC), 100 μM; D) U73122 (U73), 10 μM and E) Ryanodine (Rya), 50 μM. F) Area under the curve of insulin-induced mitochondrial Ca2+ signals of control, Ru, XeC, U73 and Rya pre-treated cardiomyocytes after insulin stimulation between 100 and 300 s. The results are representative of 3 independent experiments (N = 3), where 10-20 cells were analysed. Data are expressed as mean ± SEM, *P < 0.05 vs. Ctrl. G) Measurement of insulin-induced mitochondrial Ca2+ signal in cardiomyocytes transfected with a siRNA control (black line) or a siRNA against MCU (gray line). Insulin (100 nM) was added at 100 s. H) Area under the curve of insulin-induced mitochondrial Ca2+ signals of control and siRNA-mCU treated cardiomyocytes. The results are representative of 3 independent experiments (N = 3), where 10-20 cells were analysed. Data are expressed as mean ± SEM, **P < 0.01 vs. siRNA Ctrl.
High concentrations of Ru can bind non-specifically to phospholipid membranes [27] potentially altering intracellular Ca2+ concentrations in our experiments. To control this, we used Fura2 to assess changes in cytoplasmic Ca2+ at baseline and after the addition of CCCP (50 μM) or thapsigargin (Thapsi, 500 μM) to deplete mitochondrial or ER Ca2+, respectively in cells treated with and without Ru (10 μM) for 3 h (Additional file 2: Figure S2A-C). No significant difference was found in basal cytoplasmic Ca2+ levels of cells treated with Ru in comparison with control cells, however, there was a slight, but not significant, decrease in the rate of Ca2+ increase (slope, [(F340/F380)/Time)] evoked with both stimuli, CCCP and Thapsi. Moreover, to further confirm the involvement of MCU in our model we used a complementary approach to alter MCU activity that was independent of any nonspecific effects of Ru. Cardiomyocytes were transiently transfected with a small interfering RNA (siRNA) against MCU, reaching a knockdown of 72%, 72 h post transfection (Additional file 2: Figure S2D-E). There was a significant reduction on insulin-induced mitochondrial Ca2+ uptake in siRNA-MCU depleted cells compared to cardiomyocytes transfected with a control siRNA (Figure 1G and H). Taken together, these results identify the insulin-induced increase in mitochondrial Ca2+ as a PLC/InsP3R/MCU dependent pathway.
The Insulin-induced increase in mitochondrial Ca2+ is reduced in norepinephrine-treated hypertrophic cardiomyocytes
During pathological hypertrophy, the heart develops significant changes in energy metabolism, shifting to an insulin resistant state [4]. To test whether the insulin-induced mitochondrial Ca2+ signal described here is altered in pathological hypertrophy, we evaluated this response in cardiomyocytes pre-treated with the adrenergic agonist NE (10 μM) for 24 h to induce hypertrophy (Additional file 3: Figure S3). Insulin-dependent mitochondrial Ca2+ uptake was reduced in the NE-treated cardiomyocytes compared to control cells (Figure 2A). Importantly, the insulin-induced increase in cytoplasmic Ca2+ levels was not reduced compared to controls in the NE-treated cardiomyocytes, as evaluated using Fluo3-AM (Figure 2B). Next, IGF-1-induced hypertrophy was used as an in vitro model of physiological hypertrophy [28]. Cardiomyocytes were treated with IGF-1 for 24 h prior to insulin stimulation. The mitochondrial Ca2+ signal induced by insulin was not reduced in IGF-1-treated cardiomyocytes compared to controls (Figure 2C and D), suggesting that the changes in mitochondrial Ca2+ uptake may be unique to the NE-induced pathological hypertrophy. To determine whether the reduction in mitochondrial Ca2+ uptake observed in NE-treated cardiomyocytes was due to a change upstream or downstream of Ca2+ release from ER, histamine was used to stimulate InsP3R-dependent Ca2+ release from the ER. Mitochondrial Ca2+ uptake after histamine stimulation was also reduced in NE-treated hypertrophic cardiomyocytes compared to control cells (Figure 2E and F). These results suggest that insulin-dependent mitochondrial Ca2+ uptake is lower in NE-treated cardiomyocytes (pathological hypertrophy) compared with control conditions. Importantly, this inhibitory effect appears not to be operative in IGF-1-treated cardiomyocytes (physiological hypertrophy) and thus is not simply a requisite change in response to cell growth. Moreover, histamine-dependent mitochondrial Ca2+ uptake was decreased in NE-treated cardiomyocytes, suggesting that changes in mitochondrial Ca2+ uptake in NE-treated cardiomyocytes occur downstream of Ca2+ release from ER and are not due to an upstream change in the insulin signaling. Interestingly, Ca+2 release from the ER after Thapsi treatment is augmented in NE-treated cardiomyocytes in comparison to control and IGF-1-treated cardiomyocytes, further indicating that the reduction in mitochondrial Ca2+ uptake is not due to a reduction in ER Ca2+ loading (Additional file 4: Figure S4).

Reduction in insulin-dependent mitochondrial Ca2+uptake in NE-induced hypertrophic cardiomyocytes. Representative measures of fluorescence-associated insulin-induced Ca2+ signals in cardiomyocytes pre-treated with (gray line) or without (Ctrl, black line) the respective hypertrophic inducers for 24 h. Insulin (100 nM) was added at 100 s. A) Mitochondrial Ca2+ signals in cardiomyocytes treated with NE 10 μM for 24 h. B) Cytoplasmic Ca2+ signals in cardiomyocytes treated with NE 10 μM for 24 h. C) Mitochondrial Ca2+ signals in cardiomyocytes treated with IGF-1 100 nM for 24 h. D) Area under the curve of mitochondrial Ca2+ signals of control and IGF-1 and NE treated cardiomyocytes after insulin stimulation between 100 and 300 s. The results are representative of 3 independent experiments (N = 3), where 10–20 cells were analysed. Data are expressed as mean ± SEM, *P < 0.05 vs. Ctrl. E) Measurement of histamine-induced mitochondrial Ca2+ signal in cardiomyocytes treated with NE 10 μM for 24 h. Histamine (100 mM) was added at 100 s. F) Area under the curve of mitochondrial Ca2+ signals of control and NE treated cardiomyocytes after histamine stimulation between 100 and 300 s. The results are representative of 3 independent experiments (N = 3), where 10–20 cells were analysed. Data are expressed as mean ± SEM, *P < 0.05 vs. Ctrl.
ER-mitochondria coupling is reduced in NE-treated hypertrophic cardiomyocytes
The distance between ER and mitochondria is a key determinant of Ca2+ transfer between these organelles [29]. To evaluate whether this parameter was altered in the NE-treated cardiomyocytes, the colocalization of ER and mitochondria was measured using organelle specific fluorescent probes and confocal microscopy. Figure 3A shows representative images of control and NE or IGF-1 treated cardiomyocytes labeled with ER-Tracker red and MitoTracker green, to detect ER and mitochondria, respectively. The colocalization of these organelles was assessed in the whole cell by the quantification of Manders’ coefficient [30],[31] which is a relative measure of the percentage of one structure in contact with other. The Manders’ coefficient M1 denotes the fraction of ER that colocalizes with mitochondria (Figure 3B), whereas the Manders' coefficient M2 denotes the fraction of mitochondria that colocalizes with ER (Figure 3C). M2 was significantly reduced in NE-treated cardiomyocytes compared with controls, indicating movement of mitochondria away from sites of contact with the ER. M1 did not change. Together with the Ca2+ uptake results, this data suggest there is a decrease in both physical and functional ER-mitochondrial coupling during pathologic hypertrophy. IGF-1-treated cardiomyocytes showed no change in either coefficient, consistent with maintenance of ER-mitochondria coupling during IGF-1-induced physiological hypertrophy. To further analyse the ER-mitochondria interaction, we measured the Manders’ coefficient using an algorithm for the ImageJ software previously developed by our group that is designed to scan the cell radially from the centre of the nucleus towards the plasma membrane in a full angle (0-360°), as illustrated in the upper left hand box of Figure 3A [31]. In this analysis, the control cells showed little variation in either Manders’ coefficient across the various regions of the cell, indicating that ER-mitochondria coupling is homogeneously distributed throughout the cell. Interestingly, this homogeneous distribution was maintained after either NE or IGF-1 treatment (Figure 3D and E). The reduction in M2 observed in NE-treated cardiomyocytes was observed in perinuclear, central, and radial regions, indicating that the reduction in ER-mitochondrial coupling occurs throughout the cardiomyocyte (Figure 3E). Because we previously identified InsP3R as the primary Ca2+ channel involved in Ca2+ transfer to the mitochondria, we evaluated the subcellular distribution of the most expressed InsP3R type 2 isoform (InsP3R2) and the mitochondrial marker mtHsp70. Figure 3F and G shows a reduction in the colocalization of InsP3R2 and the mitochondrial marker mtHsp70 in NE-treated cardiomyocytes compared to vehicle treated controls. In this case, both M1 and M2 were significantly reduced. We realize that there are important differences between the ER-mitochondrial interface in a functioning heart compared to cultured cells. Therefore a comprehensive validation of the differences between the two conditions is beyond the scope of our current manuscript, and although we believe that our findings are relevant to in vivo events, clearly further experimentation will be required to validate this.

ER-mitochondria coupling is reduced in NE-induced hypertrophic cardiomyocytes. A) Representative confocal images of cardiomyocytes stained with MitoTracker green 200 nM for 30 min (Mitochondria, green) and ER-Tracker red 400 nM for 30 min (ER, red), treated with NE 10 μM or IGF-1 100 nM for 24 h. Scale bar: 10 μM. B-C) Quantification of the Manders’ coefficient M1 (fraction of ER in colocalization with mitochondria), or M2 (fraction of mitochondria in colocalization with ER). Data are expressed as mean ± SEM, N = 3, *P < 0.05 vs. Ctrl. D-E) Quantification of the M1 and M2 coefficients within the predefined subcellular regions [perinuclear (peri), central and radial]. Data are expressed as mean ± SEM, N = 3, *P < 0.05 vs. Control. F) Representative confocal images of cardiomyocytes stained for InsP3R2 (green) and mtHsp70 (red), treated with NE 10 μM for 24 h. Scale bar: 20 μM. G) Quantification of the Manders’ coefficient M1 (InsP3R2 overlapping mtHsp70) and M2 (mtHsp70 overlapping InsP3R2). Data are expressed as mean ± SEM, N = 3, **P < 0.01 vs. Ctrl. H) Oxygen consumption rate of cardiomyocytes treated with NE 10 μM or IGF-1 100 nM for 24 h. Data are expressed as mean ± SEM, N = 6, *P < 0.05 and **P < 0.01 vs. Control (ctrl).
Next, we tested if the reduction in ER-mitochondria coupling in NE-treated cardiomyocytes correlated with a change in mitochondrial metabolism. The rate of oxygen consumption was significantly reduced in NE-stimulated cardiomyocytes compared to controls. (78 ± 4, p <0.05) (Figure 3H). Oxygen consumption in IGF-1-stimulated cardiomyocytes was increased (127 ± 10, p <0.01) in comparison to control cells. All together, these data indicate that NE-induced pathologic hypertrophy causes a reduction in the physical association between ER and mitochondria and a reduction in mitochondrial metabolism, whereas IGF-1-induced physiologic hypertrophy does not. This reduction in physical contact between ER and mitochondria could be an underlying cause contributing to the reduction in insulin-stimulated mitochondrial Ca2+ uptake and mitochondrial metabolism seen under certain pathological hypertrophic states.
Insulin signaling is blunted in NE-treated cardiomyocytes
Pathological hypertrophy is associated with a decline in insulin sensitivity in both animal models and clinical populations with hypertrophic heart disease [20],[21]. To determine whether canonical insulin signaling was altered in our in vitro cell culture model of NE-induced hypertrophy, various cellular processes regulated by insulin were measured. Akt is a serine/threonine kinase downstream of insulin receptor whose activity is regulated by phosphorylation [32]. Insulin treatment of control cardiomyocytes induced phosphorylation of Akt at Ser473 (Figure 4A). This response was reduced in hypertrophic cardiomyocytes pre-treated with NE, indicating a blunting of this early effector of insulin signaling. Consistent with this finding, insulin-stimulated glucose uptake was also reduced in the NE-treated cardiomyocytes (Figure 4B).

Hypertrophic cardiomyocytes show a reduced insulin response. A) Western blot of p-Akt (Ser473) and Akt (Upper) and densitometric analysis (lower) of control and hypertrophic NE-treated cardiomyocytes (NE 10 μM, 24 h) either stimulated with insulin 100 nM for 15 min or left unstimulated. Data were relativized against insulin (white bar) and expressed as mean ± SEM, N = 6, ***P < 0.001 vs. insulin. B) Glucose uptake of control and hypertrophic NE-treated cardiomyocytes (NE 10 μM for 24 h) either stimulated with insulin 100 nM for 15 min or left unstimulated. Cytochalasin B (Cyto B) was used as a negative control. Data are expressed as mean ± SEM, N = 7, **P < 0.01, ns: non significant. C) Oxygen consumption rates of control and hypertrophic cardiomyocytes (NE 10 μM for 24 h) either stimulated with insulin 100 nM for 3 h or left unstimulated. Data are expressed as mean ± SEM, N = 3, **P < 0.01 vs. control, ns: non significant. D) Mitochondrial membrane potential quantification in control cardiomyocytes or treated with NE 10 μM for 24 h and stimulated with insulin. CCCP (50 μM, 30 min) was used as a negative control. Data are expressed as mean ± SEM, N = 3, *P < 0.05 vs. control, ns: non significant. E) qPCR for Hk2, Pfkfb2, Slc2a1, Slc2a4 and Pdk4 mRNA levels of control (Ctrl) and hypertrophic cardiomyocytes (NE 10 μM for 24 h) either stimulated with insulin 100 nM for 3 h or left unstimulated. Data are expressed as mean ± SEM, N = 4, *P < 0.05, **P < 0.01 and ***P < 0.001 vs. Ctrl; ###P < 0.001 vs. Insulin and &&&P < 0.001 vs. NE.
Previously, we and other groups demonstrated that insulin induces a rise in mitochondrial metabolism, that is fundamental to its ability to act as a metabolic regulator [25],[33],[34]. Therefore, we evaluated changes in mitochondrial respiration 3 h after insulin treatment by measuring oxygen consumption and mitochondrial membrane potential. Control cardiomyocytes showed a significant increase in oxygen consumption in response to insulin stimulation (134% ±8, p <0.01), whereas insulin did not significantly increase oxygen consumption in the NE-treated cardiomyocytes (Figure 4C). In parallel, mitochondrial membrane potential measured by flow cytometry, showed a very similar behaviour. Figure 4D shows that insulin increased mitochondrial membrane potential (138% ±10, p <0.05), whereas pre-treatment with NE completely abolished this response. Together, these results confirm that the metabolic response to insulin is reduced in NE-treated cardiomyocytes, consistent with reductions in insulin sensitivity reported for pathological hypertrophy in vivo. Because pyruvate dehydrogenase (PDH)-dependent glycolysis can be up-regulated in hypertrophy, we also analyzed the expression of five glycolytic genes (glycolytic markers): hexokinase 2 (Hk2), 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 2 (Pfkfb2), Glut1 (Slc2a1), Glut4 (Slc2a4) and pyruvate dehydrogenase kinase, isozyme 4 (Pdk4). Figure 4E shows that expression of Hk2, Pfkfb2, Slc2a1 and Slca2a4 increase additively with insulin and NE treatments. However, in the case of Pdk4, a key enzyme down-regulated by insulin, NE had the opposite effect, increasing expression as well as preventing insulin-mediated downregulation. These data point to PDK4 regulation of PDH as a potentially interesting point of regulation between the glycolytic induction triggered by insulin and the chronic effects exerted by NE during hypertrophy.
Blocking mitochondrial Ca2+ uptake mimics the hypertrophic metabolic phenotype
To test for a causative relationship between changes in mitochondrial Ca2+ uptake and changes in insulin sensitivity, we pre-treated cardiomyocytes with Ru for 30 min to inhibit Ca2+ entry into mitochondria [35] and then evaluated the insulin-induced metabolic response of the cells. The response of Ru-treated cardiomyocytes was a phenotypic copy of NE-treated cardiomyocytes (Figure 4). Insulin-induced Ser473-Akt phosphorylation was reduced in cardiomyocytes with either Ru-treatment or siRNA-MCU knockdown (Figure 5A and B). Insulin-induced glucose uptake and stimulation of oxygen consumption were likewise reduced (Figure 5C-D). Furthermore, the insulin-induced increase in the oxygen consumption in cardiomyocytes was also blocked by pre-treatment with the InsP3R-specific antagonist XeC (which blocks Ca2+ release from the ER) as well as by depletion of MCU with siRNA (Figure 5D). Figure 5E shows the evaluation of the glycolytic genes assessed previously in Figure 4E. Consistent with the preceding data, insulin, Ru and XeC increased expression of the first four markers, whereas, Ru and XeC overcame the inhibitory effect of insulin on Pdk4 expression.

The blockade of Ca2+entry to the mitochondria reduces insulin response. A) Western blot of p-Akt (Ser473) and Akt (upper) and densitometric analysis (lower) of cardiomyocytes treated with ruthenium red (Ru 10 μM for 30 min) either stimulated with insulin (100 nM for 15 min) or left unstimulated. Data are expressed as mean ± SEM, N = 6, *P < 0.05 vs. insulin. B) Western blot of p-Akt (Ser473) and Akt (upper) and densitometric analysis (lower) of cardiomyocytes transfected with a siRNA Ctrl or against MCU either stimulated with insulin (100 nM for 15 min) or left unstimulated. Data are expressed as mean ± SEM, N = 3, *P < 0.05 vs. insulin. C) Glucose uptake of cardiomyocytes treated with ruthenium red (Ru 10 μM for 30 min) either stimulated with insulin 100 nM for 15 min or left unstimulated. Cytochalasin B (Cyto B) was used as a negative control. Data are expressed as mean ± SEM, N = 7, **P < 0.01 vs. control, ns: non significant. D) Oxygen consumption rates of cardiomyocytes treated with ruthenium red (Ru 10 μM, 30 min prior stimuli) or xestospongin C (XeC, 100 μM, 30 min prior stimuli) either stimulated with insulin (100 nM for 3 h) or left unstimulated. Data are expressed as mean ± SEM, N = 3, **P < 0.01 vs. control ns: non significant. E) qPCR for Hk2, Pfkfb2, Slc2a1, Slc2a4 and Pdk4 mRNA levels of cardiomyocytes treated with ruthenium red (Ru 10 μM for 30 min) or xestospongin C (XeC, 100 mM, 30 min prior stimuli) either stimulated with insulin (100 nM for 15 min) or left unstimulated. Data are expressed as mean ± SEM, N = 4, *P < 0.05 vs. Ctrl and #P < 0.05 vs. Insulin.
Finally, to better understand the mechanism through which NE and mitochondrial Ca2+ uptake may be cross talk with insulin and Akt signalling, we assessed changes in phosphorylation of IRS-1 at Ser307. Phosphorylation of this site is inhibitory, uncoupling IRS-1 from the insulin receptor and decreasing activation of downstream targets including Akt [36]. A number of pathways can lead to Ser307 phosphorylation including insulin stimulation itself, thus providing feed back control over insulin stimulation. Consistent with decrease insulin-sensitivity in pathological hypertrophy [32] and the findings in Figure 4A, NE decreased insulin-induced phosphorylation of IRS-1 (Additional file 5: Figure S5). In this instance, the effect of Ru and XeC on insulin-stimulated Ser307 phosphorylation was less pronounced and not significant, suggesting that the impact of mitochondrial Ca2+ uptake on Akt signalling may not be the direct consequence of changes in insulin receptor sensitivity. This also highlights the complexity of the signalling mechanisms involved that merit further investigation.
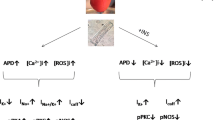
Taken together, these data suggest that Ca2+ entry into mitochondria is an important aspect of the heart;s response to insulin, modulating both the activity of intermediates and the final metabolic effects at the level of cardiomyocytes. Furthermore, we show that this pathway of intracellular communication is altered in pathological, but not physiological hypertrophy and speculate that changes in this process could underlie the reduced insulin responses observed in other pathologies. The possible retrograde modulation of mitochondria to insulin signaling reveals an important new aspect of cellular crosstalk, placing mitochondria as an active participant in intracellular communication (Figure 6).

Proposed model for insulin-induced mitochondrial Ca2+uptake in normal and hypertrophic cardiomyocytes. During hypertrophy there is a decrease in ER-mitochondria physical coupling, disrupting Ca2+ transfers between both organelles, thus leading a diminished Akt phosphorylation and oxygen consumption in response to insulin.
Discussion
In recent decades there have been important advances in our understanding of the pathways mediating insulin signaling. The most widely studied is the canonical signaling cascade constituted by PI3K/Akt, that controls glucose uptake, metabolic activity, and translational responses to insulin [37]. Ca2+ has also been identified as an important second messenger acting downstream of the insulin receptor and shown to be essential in insulin-mediated glucose uptake in cardiac and skeletal muscle [38]-[40]. Our group showed that Ca2+ release from the ER through the InsP3R is an important component of the Ca2+-mediated insulin response in cardiomyocytes [19]. Mitochondria have a substantial capacity for Ca2+ storage that is facilitated by their physical and functional interaction with InsP3Rs in the ER membrane [41]. As such, poor or excessive mitochondrial Ca2+ retention represent a potential mechanism contributing to metabolic imbalance and insulin resistance.
The work presented here is the first to identify mitochondrial Ca2+ uptake as a key event in insulin-mediated signalling pathways in cardiomyocytes. We show that insulin-induced stimulation of oxidative metabolism via mitochondrial uptake of Ca2+ released from ER, whereas it is diminished in norepinephrine-treated cardiomyocytes undergoing pathological hypertrophy (Figure 6). Our results highlight the involvement of two intracellular Ca2+ channels: InsP3R, a principal Ca2+ ER channel, and MCU, which carries out mitochondrial Ca2+ uptake. The proximity of these two channels determines the efficiency of transfer of Ca2+ from ER to mitochondria.
Mitochondrial insulin-dependent Ca2+ signals in hypertrophic cardiomyocytes
Mobilization of Ca2+ is known to facilitate insulin-stimulated glucose uptake in diverse cell types [38]-[40]. However, the precise mechanisms of action, the specific pools of Ca2+ involved, and how these phenomena may change under pathological conditions are largely unknown. Here, we evaluated both cytoplasmic and mitochondrial insulin-dependent Ca2+ signals in cardiomyocytes and assessed whether signaling is altered in hypertrophy. Deregulation of cytoplasmic Ca2+ levels and changes in the expression of various Ca2+ channels are hallmarks of cardiac hypertrophy [42]-[44]. However, with regards to cytoplasmic Ca2+ signals induced by insulin we found no alterations in the shape, timing or intensity in hypertrophic cardiomyocytes. This result, although surprising, does not preclude the possibility of alterations specific to particular cellular regions. In contrast, the mitochondrial Ca2+ response was blunted specifically in NE-treated cardiomyocytes, a model of pathological hypertrophy, but not in cardiomyocytes exposed to IGF-1, as a model of physiological hypertrophy (Figure 2). Mitochondrial Ca2+ uptake in response to histamine-mediated release of Ca2+ from the ER was likewise reduced in NE-treated cardiomyocytes compared to control or IGF-1 treated cardiomyocytes. This result demonstrates that the observed effects are not particular to insulin, but instead indicate a general decrease in the capacity for mitochondrial Ca2+ uptake in pathological hypertrophy. Our data are consistent with the work of Fauconnier et al. which described alterations in cytoplasmic and mitochondrial Ca2+ signaling in response to electrical stimulation in cardiomyocytes obtained from obese mice, another pathological condition where the heart develops insulin resistance [45]. In their study, using adult cardiomyocytes isolated from wild type and ob/ob mice, electrically evoked mitochondrial Ca2+ uptake was increased by insulin in cells from wild type, but not ob/ob. These findings suggest that, in addition to direct stimulation of mitochondrial Ca2+ uptake, insulin can increase the magnitude of uptake induced by other stimuli.
Our finding that blocking the entry of Ca2+ to mitochondria with Ru decreases insulin-dependent Akt phosphorylation suggests that this Ca2+ dynamic is relevant for proper insulin signaling to Akt. In this context, our group has previously demonstrated that inhibition of InsP3R with XeC and the Ca2+ chelating agent BAPTA-AM also reduces Akt phosphorylation [19]. Moreover, we showed that the induction of mitochondrial fragmentation reduces insulin-induced Akt phosphorylation by reducing mitochondrial Ca2+ uptake [35]. However, further investigation will be needed to fully understand the mechanisms involved in this signalling cross-talk. Taken together, these findings suggest that a reduction in the capacity of mitochondria to uptake Ca2+ may be a common feature of insulin resistance.
ER-mitochondrial coupling in hypertrophic cardiomyocytes
Although the mechanism and regulatory components involved in mitochondrial Ca2+ uptake after ER release have been described, alterations in this process in pathological conditions are still unknown. The distance between ER and mitochondria is a key factor that determines the efficiency of Ca2+ transfer [46]. The ER and mitochondrial networks are highly dynamic and can change their shape and/or distribution in response to various stimuli. Bravo et al. showed that in early stages of ER stress, both mitochondria and ER migrate to the perinuclear region of the cell, increasing contacts between both organelles, enhancing Ca2+ transfer. This ultimately leads to an increase in mitochondrial metabolism [31]. We tested whether changes in ER-mitochondria coupling in pathological hypertrophy could underlie the observed decrease in insulin-stimulated mitochondrial Ca2+ uptake. Indeed, Figure 3 shows decreased colocalization and ER-mitochondrial coupling in pathological (NE) hypertrophied cardiomyocytes as compared to control or physiological (IGF-1) hypertrophied cardiomyocytes. The increased distance between the two-organelle populations would increase the potential for dispersion of Ca2+ released from the ER and prevent local Ca2+ concentrations at the ER-mitochondrial interface from reaching the critical concentration required for mitochondrial Ca2+ uptake via the MCU.
Our data show that reducing mitochondrial Ca2+ uptake with Ru is sufficient to confer an insulin-resistant phenotype with regards to Akt activation, glucose uptake, and oxygen consumption. Thus, mitochondrial Ca2+ uptake is not only a downstream target of insulin signaling, it also appears to participate in feed-forward signaling that helps to maintain insulin sensitivity.
Conclusions
Mitochondrial Ca2+ uptake is a key event in insulin signalling and metabolism in cardiomyocytes. Our findings contribute to a better understanding of the causes that trigger insulin resistance in pathological hypertrophy. This work raises important new questions that place mitochondrial Ca2+ handling at a nodal point of metabolic control and demonstrate that preservation of this function is essential for proper cardiac function.
Methods
Culture and treatment of cardiomyocytes
Neonatal ventricular cardiomyocytes were prepared from hearts of 1-3-day-old Sprague Dawley rats as described previously [47]. They were euthanized by decapitation. Ventricles were trisected, pooled, and cardiomyocytes dissociated in a solution of collagenase and pancreatin. After enzymatic dissociation, cells were selectively enriched for cardiomyocytes by being pre-plated in DMEM/M199 (4:1) containing 10% (v/v) foetal bovine serum, 5% (v/v) foetal calf serum, penicillin, and streptomycin (100 units/ml). Cardiomyocytes were plated at a density of 2.5x105 or 1.5x106 for microscopy or Western blot experiments respectively. Serum was withdrawn for 24 h before the cells were treated with insulin (100 nM) and mitochondrial and cytoplasmic Ca2+ were assessed. In some experiments, cardiomyocyte hypertrophy was induced with NE (10 μM) or IGF-1 (100 nM) for 24 h and then stimulated with insulin. To inhibit the mitochondrial Ca2+ uniporter, Ru and siRNA to MCU were used. The InsP3R, ryanodine receptor and PLC were inhibited with XeC, ryanodine and U73122, respectively. Studies were approved by the Institutional Bioethical Committee, Faculty of Chemical and Pharmaceutical Sciences, University of Chile, in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals published by the US (NIH Publication, 8th Edition, 2011).
Cardiomyocyte transfection
Small interfering RNAs (siRNAs) for MCU I and II and negative control (MISSION; Sigma-Aldrich Co.) were used according to manufacturer's instructions. The siRNAs used for knockdown experiments were as follows: negative control, catalogue number SIC001; MCU I, sense (5'-CAGAGACCCUGAACGAUGU-3'), antisense (5'-ACAUCGUUCAGGGUCUCUG-3'); and MCU II, sense (5'-GGCUUACCUGGUGGGAAUA-3'), antisense (5'-UAUUCCCACCAGGUAAGCC-3'). Both siRNAs were tested, obtaining the best results with the siRNA that we called II, which was the one used for all the subsequent experiments (Additional file 2: Figure S2).
Intracellular and mitochondrial Ca2+ determinations
Cytosolic Ca2+ levels were determined in cardiomyocytes preloaded with Fluo3-AM (5.4 μM, 30 min) or Fura2 (5 μM, 30 min), as described previously [19],[48]. To determine mitochondrial Ca2+ levels, images were obtained from cultured cardiomyocytes preloaded with Rhod-FF (5.4 μM, 30 min) [49],[50]. At the end of each measurement, CCCP 10 μM was used as control. Both determinations were performed in an inverted confocal microscope (Carl Zeiss LSM-5, Pascal 5 Axiovert 200 microscope).
ER and mitochondrial network imaging
For ER and mitochondrial imaging, cells were treated during 30 min with 200 nM ER-Tracker red and with 400 nM MitoTracker Green, respectively. Confocal image stacks were captured with an inverted confocal microscope (Carl Zeiss LSM-5, Pascal 5 Axiovert 200 microscope).
Image processing
For mitochondrial network and ER colocalization, one focal plane was analyzed. The images obtained were deconvolved using the ImageJ software. Colocalization between organelles was quantified using the Manders’ algorithm, as previously described [24],[30],[31],[51]. For each independent experiment, 10-15 cells were registered.
Mitochondrial dynamics analysis
Cells were incubated for 30 min with MitoTracker Green FM (400 nM) with or without Rhod-FF (5.4 μM) and maintained in Krebs solution. Confocal image stacks were captured with a Leica TCS SP5 confocal microscope and a Plan-APOCHROMAT 63x/1.4 Oil DIC objective as previously described [24],[25]. Images were deconvolved with ImageJ software (National Institutes of Health), and volume reconstitution of Z-stacks of thresholded images was performed. The number and individual volume of each object (mitochondria) were quantified with the ImageJ 3D Object Counter plug-in. Each experiment was done at least three times, and 16-25 cells per condition were quantified.
Western blot
The primary antibodies used were anti-β-MHC (1:1000, Sigma-Aldrich), anti-phospho-Akt (Ser473) (1:1000, Cell Signaling), anti-Akt (1:1000, Cell Signaling), anti IRS-1 (1:1000, Cell Signaling), anti-phospho-IRS-1 (Ser307) (1:500, Cell Signaling) and anti-MCU (1:2000, Abcam). Then the blots were incubated with a horseradish peroxidase-coupled secondary antibody (1:5000, Pierce). ImageJ software was used for image densitometry.
Oxygen consumption
Cardiomyocytes were plated and treated according each experiment. Cells were then trypsinized, and the suspension was placed in a chamber at 25°C, coupled to a Clark electrode 5331 (Yellow Springs Instruments) where the oxygen consumption was measured polarographically [31],[50].
Glucose uptake
Cardiomyocytes were rinsed twice with HEPES-buffered saline. Inhibitors were added for 30 min and insulin during the last 10 min. Glucose uptake was measured using 10 mM [3H]2-deoxyglucose [19].
Flow cytometry analysis of mitochondrial membrane potential
ΔΨM was measured after loading cardiomyocytes with TMRM (200 nM) for 30 min. Afterward, cells underwent trypsinization, and fluorescence was assessed by flow cytometry (excitation/emission 543/560) with a FACScan system (Becton Dickinson). CCCP (50 mM) and oligomycin (10 mM) for 30 min were used as positive and negative controls for the ΔΨM measurements.
qPCR
Real-time PCR was performed with SYBR green (Applied Biosystems) as previously described [25]. Data for each transcript was normalized to 18S rRNA as internal control with the 2-ΔΔCt method. Primers used were as follows: MCU rat forward 5'-GCGCCAGGAATATGTTTATC-3'; MCU rat reverse 5'-TTGCATCCTTGAGTTGATTG-3'; Hk2 rat forward 5'-CGAATCAAAGAGAACAAGGG-3'; Hk2 rat reverse 5'-CAAAATGGGGATGTTTCTTG-3'; Pfkfb2 rat forward 5'-CTGGAGGTAAAAGTGTCAAG-3'; Pfkfb2 rat reverse 5'-ACGAGAGGTCCTTATCATAG-3'; Slc2a1 rat forward 5'-CAATATGTGGAGCAACTGTG-3'; Slc2a1 rat reverse 5'-AGTAGGTGAAGATGAAGAAGAG-3'; Slc2a4 rat forward 5'-AAGTGATTGAACAGAGCTAC-3'; Slc2a4 rat reverse 5'-CTTTTCCTTCCCAACCATTG-3'; Pdk4 rat forward 5'-AATCAAGATTTCTGACCGAG-3'; Pdk4 rat reverse 5'-CTGACATGGAATAGAGATTCAG-3'.
Statistical analysis
Data are presented either as means ± SEM of a number (n) of independent experiments or as examples of representative experiments performed on at least three separate occasions. Data were analysed by analysis of variance and comparisons between groups were performed using a protected Tukey t test. A value of P < 0.05 was chosen as the limit of statistical significance.
Additional files
References
Hill JA, Olson EN: Cardiac plasticity. N Engl J Med. 2008, 358: 1370-1380. 10.1056/NEJMra072139.
Allard MF, Schönekess BO, Henning SL, English DR, Lopaschuk GD: Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol. 1994, 267: H742-H750.
Kolwicz SC, Tian R: Glucose metabolism and cardiac hypertrophy. Cardiovasc Res. 2011, 90: 194-201. 10.1093/cvr/cvr071.
Ingwall JS: Energy metabolism in heart failure and remodelling. Cardiovasc Res. 2009, 81: 412-419. 10.1093/cvr/cvn301.
Razeghi P, Young ME, Alcorn JL, Moravec CS, Frazier OH, Taegtmeyer H: Metabolic gene expression in fetal and failing human heart. Circulation. 2001, 104: 2923-2931. 10.1161/hc4901.100526.
Heineke J, Ritter O: Cardiomyocyte calcineurin signaling in subcellular domains: from the sarcolemma to the nucleus and beyond. J Mol Cell Cardiol. 2012, 52: 62-73. 10.1016/j.yjmcc.2011.10.018.
Anderson ME, Brown JH, Bers DM: CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol. 2011, 51: 468-473. 10.1016/j.yjmcc.2011.01.012.
Liu Q, Molkentin JD: Protein kinase Cα as a heart failure therapeutic target. J Mol Cell Cardiol. 2011, 51: 474-478. 10.1016/j.yjmcc.2010.10.004.
Cárdenas C, Miller RA, Smith I, Bui T, Molgó J, Müller M, Vais H, Cheung K-H, Yang J, Parker I, Thompson CB, Birnbaum MJ, Hallows KR, Foskett JK: Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010, 142: 270-283. 10.1016/j.cell.2010.06.007.
Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R: Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc Natl Acad Sci USA. 1999, 96: 13807-13812. 10.1073/pnas.96.24.13807.
Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK: Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011, 476: 341-345. 10.1038/nature10234.
Santo-Domingo J, Demaurex N: Calcium uptake mechanisms of mitochondria. Biochim Biophys Acta. 2010, 1797: 907-912. 10.1016/j.bbabio.2010.01.005.
Csordás G, Várnai P, Golenár T, Roy S, Purkins G, Schneider TG, Balla T, Hajnóczky G: Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol Cell. 2010, 39: 121-132. 10.1016/j.molcel.2010.06.029.
Giacomello M, Drago I, Bortolozzi M, Scorzeto M, Gianelle A, Pizzo P, Pozzan T: Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Mol Cell. 2010, 38: 280-290. 10.1016/j.molcel.2010.04.003.
Hayashi T, Rizzuto R, Hajn°Czky G, Su T-P: MAM: more than just a housekeeper. Trends Cell Biol. 2009, 19: 81-88. 10.1016/j.tcb.2008.12.002.
Bravo R, Parra V, Gatica D, Rodriguez AE, Torrealba N, Paredes F, Wang ZV, Zorzano A, Hill JA, Jaimovich E, Quest AFG, Lavandero S: Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. Int Rev Cell Mol Biol. 2013, 301: 215-290. 10.1016/B978-0-12-407704-1.00005-1.
Pirola L, Johnston AM, Van Obberghen E: Modulation of insulin action. Diabetologia. 2004, 47: 170-184. 10.1007/s00125-003-1313-3.
Brownsey RW, Boone AN, Allard MF: Actions of insulin on the mammalian heart: metabolism, pathology and biochemical mechanisms. Cardiovasc Res. 1997, 34: 3-24. 10.1016/S0008-6363(97)00051-5.
Contreras-Ferrat AE, Toro B, Bravo R, Parra V, Vásquez C, Ibarra C, Mears D, Chiong M, Jaimovich E, Klip A, Lavandero S: An inositol 1,4,5-triphosphate (IP3)-IP3 receptor pathway is required for insulin-stimulated glucose transporter 4 translocation and glucose uptake in cardiomyocytes. Endocrinology. 2010, 151: 4665-4677. 10.1210/en.2010-0116.
Paolisso G, Galzerano D, Gambardella A, Varricchio G, Saccomanno F, D'Amore A, Varricchio M, D'Onofrio F: Left ventricular hypertrophy is associated with a stronger impairment of non-oxidative glucose metabolism in hypertensive patients. Eur J Clin Invest. 1995, 25: 529-533. 10.1111/j.1365-2362.1995.tb01740.x.
Paternostro G, Clarke K, Heath J, Seymour AM, Radda GK: Decreased GLUT-4 mRNA content and insulin-sensitive deoxyglucose uptake show insulin resistance in the hypertensive rat heart. Cardiovasc Res. 1995, 30: 205-211. 10.1016/S0008-6363(95)00019-4.
Friehs I, Moran AM, Stamm C, Colan SD, Takeuchi K, Cao-Danh H, Rader CM, McGowan FX, del Nido PJ: Impaired glucose transporter activity in pressure-overload hypertrophy is an early indicator of progression to failure. Circulation. 1999, 100: II187-II193. 10.1161/01.CIR.100.suppl_2.II-187.
Fonteriz RI, Moreno A, Lobatón CD, Montero M, Alvarez J: Monitoring mitochondrial [Ca(2+)] dynamics with rhod-2, ratiometric pericam and aequorin. Cell Calcium. 2010, 48: 61-69. 10.1016/j.ceca.2010.07.001.
Parra V, Eisner V, Chiong M, Criollo A, Moraga F, Garcia A, Härtel S, Jaimovich E, Zorzano A, Hidalgo C, Lavandero S: Changes in mitochondrial dynamics during ceramide-induced cardiomyocyte early apoptosis. Cardiovasc Res. 2008, 77: 387-397. 10.1093/cvr/cvm029.
Parra V, Verdejo HE, Iglewski M, del Campo A, Troncoso R, Jones D, Zhu Y, Kuzmicic J, Pennanen C, López-Crisosto C, Jaña F, Ferreira J, Noguera E, Chiong M, Bernlohr DA, Klip A, Hill JA, Rothermel BA, Abel ED, Zorzano A, Lavandero S: Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the Akt-mTOR-NFκB-Opa-1 signaling pathway. Diabetes. 2014, 63: 75-88. 10.2337/db13-0340.
Drago I, De Stefani D, Rizzuto R, Pozzan T: Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc Natl Acad Sci. 2012, 109: 12986-12991. 10.1073/pnas.1210718109.
Voelker D, Smejtek P: Adsorption of ruthenium red to phospholipid membranes. Biophys J. 1996, 70: 818-830. 10.1016/S0006-3495(96)79621-2.
McMullen JR, Izumo S: Role of the insulin-like growth factor 1 (IGF1)/phosphoinositide-3-kinase (PI3K) pathway mediating physiological cardiac hypertrophy. Novartis Found Symp. 2006, 274: 90-111. 10.1002/0470029331.ch7. discussion 111-7- 152-5- 272-6
Giorgi C, De Stefani D, Bononi A, Rizzuto R, Pinton P: Structural and functional link between the mitochondrial network and the endoplasmic reticulum. Int J Biochem Cell Biol. 2009, 41: 1817-1827. 10.1016/j.biocel.2009.04.010.
Manders E, Verbeek FJ, Aten JA: Measurement of co-localization of objects in dual-colour confocal images. J Microsc. 1993, 169: 375-382. 10.1111/j.1365-2818.1993.tb03313.x.
Bravo R, Vicencio JM, Parra V, Troncoso R, Munoz JP, Bui M, Quiroga C, Rodriguez AE, Verdejo HE, Ferreira J, Iglewski M, Chiong M, Simmen T, Zorzano A, Hill JA, Rothermel BA, Szabadkai G, Lavandero S: Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J Cell Sci. 2011, 124: 2143-2152. 10.1242/jcs.080762.
Muniyappa R, Montagnani M, Koh KK, Quon MJ: Cardiovascular actions of insulin. Endocr Rev. 2007, 28: 463-491. 10.1210/er.2007-0006.
Cheng Z, Guo S, Copps K, Dong X, Kollipara R, Rodgers JT, Depinho RA, Puigserver P, White MF: Foxo1 integrates insulin signaling with mitochondrial function in the liver. Nat Med. 2009, 15: 1307-1311. 10.1038/nm.2049.
Liu S, Okada T, Assmann A, Soto J, Liew CW, Bugger H, Shirihai OS, Abel ED, Kulkarni RN: Insulin signaling regulates mitochondrial function in pancreatic beta-cells. PLoS ONE. 2009, 4: e7983-10.1371/journal.pone.0007983.
del Campo A, Parra V, Vásquez-Trincado C, Gutierrez T, Morales PE, López-Crisosto C, Bravo-Sagua R, Navarro-Marquez MF, Verdejo HE, Contreras-Ferrat A, Troncoso R, Chiong M, Lavandero S: Mitochondrial fragmentation impairs insulin-dependent glucose uptake by modulating Akt activity through mitochondrial Ca2+ uptake. Am J Physiol Endocrinol Metab. 2014, 306: E1-E13. 10.1152/ajpendo.00146.2013.
Rui L, Aguirre V, Kim JK, Shulman GI, Lee A, Corbould A, Dunaif A, White MF: Insulin/IGF-1 and TNF-α stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J Clin Invest. 2001, 107: 181-189. 10.1172/JCI10934.
Taniguchi CM, Emanuelli B, Kahn CR: Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006, 7: 85-96. 10.1038/nrm1837.
Whitehead JP, Molero JC, Clark S, Martin S, Meneilly G, James DE: The role of Ca2+ in insulin-stimulated glucose transport in 3'T3-L1 cells. J Biol Chem. 2001, 276: 27816-27824. 10.1074/jbc.M011590200.
Lanner JT, Katz A, Tavi P, Sandström ME, Zhang S-J, Wretman C, James S, Fauconnier J, Lännergren J, Bruton JD, Westerblad H: The role of Ca2+ influx for insulin-mediated glucose uptake in skeletal muscle. Diabetes. 2006, 55: 2077-2083. 10.2337/db05-1613.
Lanner JT, Bruton JD, Assefaw-Redda Y, Andronache Z, Zhang S-J, Severa D, Zhang Z-B, Melzer W, Zhang S-L, Katz A, Westerblad H: Knockdown of TRPC3 with siRNA coupled to carbon nanotubes results in decreased insulin-mediated glucose uptake in adult skeletal muscle cells. FASEB J. 2009, 23: 1728-1738. 10.1096/fj.08-116814.
Lehninger AL, Rossi CS, Greenawalt JW: Respiration-dependent accumulation of inorganic phosphate and Ca ions by rat liver mitochondria. Biochem Biophys Res Commun. 1963, 10: 444-448. 10.1016/0006-291X(63)90377-2.
Anger M, Lompré AM, Vallot O, Marotte F, Rappaport L, Samuel JL: Cellular distribution of Ca2+ pumps and Ca2+ release channels in rat cardiac hypertrophy induced by aortic stenosis. Circulation. 1998, 98: 2477-2486. 10.1161/01.CIR.98.22.2477.
Yatani A, Honda R, Tymitz KM, Lalli MJ, Molkentin JD: Enhanced Ca2+ channel currents in cardiac hypertrophy induced by activation of calcineurin-dependent pathway. J Mol Cell Cardiol. 2001, 33: 249-259. 10.1006/jmcc.2000.1296.
Zwadlo C, Borlak J: Disease-associated changes in the expression of ion channels, ion receptors, ion exchangers and Ca(2+)-handling proteins in heart hypertrophy. Toxicol Appl Pharmacol. 2005, 207: 244-256. 10.1016/j.taap.2005.01.005.
Fauconnier J, Lanner JT, Zhang S-J, Tavi P, Bruton JD, Katz A, Westerblad H: Insulin and inositol 1,4,5-trisphosphate trigger abnormal cytosolic Ca2+ transients and reveal mitochondrial Ca2+ handling defects in cardiomyocytes of ob/ob mice. Diabetes. 2005, 54: 2375-2381. 10.2337/diabetes.54.8.2375.
Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T: Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998, 280: 1763-1766. 10.1126/science.280.5370.1763.
Foncea R, Andersson M, Ketterman A, Blakesley V, Sapag-Hagar M, Sugden PH, LeRoith D, Lavandero S: Insulin-like growth factor-I rapidly activates multiple signal transduction pathways in cultured rat cardiac myocytes. J Biol Chem. 1997, 272: 19115-19124. 10.1074/jbc.272.31.19115.
Chiong M, Parra V, Eisner V, Ibarra C, Maldonado C, Criollo A, Bravo R, Quiroga C, Contreras A, Vicencio JM, Cea P, Bucarey JL, Molgó J, Jaimovich E, Hidalgo C, Kroemer G, Lavandero S: Parallel activation of Ca(2+)-induced survival and death pathways in cardiomyocytes by sorbitol-induced hyperosmotic stress. Apoptosis. 2010, 15: 887-903. 10.1007/s10495-010-0505-9.
Rojas-Rivera D, Díaz-Elizondo J, Parra V, Salas D, Contreras A, Toro B, Chiong M, Olea-Azar C, Lavandero S: Regulatory volume decrease in cardiomyocytes is modulated by calcium influx and reactive oxygen species. FEBS Lett. 2009, 583: 3485-3492. 10.1016/j.febslet.2009.10.003.
Troncoso R, Vicencio JM, Parra V, Nemchenko A, Kawashima Y, del Campo A, Toro B, Battiprolu PK, Aranguiz P, Chiong M, Yakar S, Gillette TG, Hill JA, Abel ED, LeRoith D, Lavandero S: Energy-preserving effects of IGF-1 antagonize starvation-induced cardiac autophagy. Cardiovasc Res. 2012, 93: 320-329. 10.1093/cvr/cvr321.
Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S: Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J. 2004, 86: 3993-4003. 10.1529/biophysj.103.038422.
Acknowledgments
This work was supported by FONDECYT (grant 1120212 to S.L., 11130285 to R.T., 11130267 to A.C-F., 1110180 and 1140329 to M.Ch., 3130749 to C.P.), CONICYT (grant FONDAP 15130011 to S.L., M.Ch., and R.T.; Red 120003 to S.L. and B.A.H.). National Institutes of Health: USA, HL072016, HL097768 (B.A.R.). We thank Becas Chile and the American Heart Association for the postdoctoral funding to V.P. and CONICYT-Chile for the Doctoral and Master fellowships to C.V.T., C.L.C. and P.M., respectively. We finally thank to Fidel Albornoz and Gindra Latorre for their excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
TG, VP, RT, CP, ACF, CVT, PEM, CLC, CSF performed all the experiments. TG, VP, RT, MC, BAH and SL wrote the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12964_2014_68_MOESM1_ESM.pdf
Additional file 1: Figure S1.: Measurement of Rhod-FF fluorescence for Ca2+ signalling in cardiomyocytes. A) Representative confocal images stained with MitoTracker green (green, top left) and Rhod-FF (red, top right). The merge between MitoTracker green and Rhod-FF is shown in the down left corner and the intensity correlation analysis and the Pearson coefficient is shown in the down right corner. Scale bar: 10 μM. B) Representative measurement of Rhod-FF fluorescence representing the insulin-induced mitochondrial Ca2+ signal. Insulin (100 nM) was added at 100 s, and CCCP (50 μM) was added at 350 s. C) Representative confocal images of cells loaded with MitoTracker Green (400 nM for 30 min) alone, or MitoTracker Green (400 nM for 30 min) and Rhod-FF (5.4 μM for 30 min). Scale bar represents 10 μM. D) Number of mitochondria per cell and individual mitochondrial volume were determined for the cells in C). Data are expressed as mean ± SEM (n = 3). (PDF 1 MB)
12964_2014_68_MOESM2_ESM.pdf
Additional file 2: Figure S2.: Ruthenium Red (Ru) and MCU siRNA knockdown controls. A) Representative measures of Fura2 fluorescence-associated CCCP (50 μM) or thapsigargin (Thapsi, 500 μM) -induced cytosolic Ca2+ signals in cardiomyocytes pre-treated with (red line) or without (Ctrl, black line) Ruthenium Red (Ru, 10 μM) for 3 h. B) Basal Fura2 Fluorescence previous to CCCP or Thapsi stimuli. C) Initial Ca2+-related fluorescence rate (slope) of control and Ru (10 μM) pre-treated cardiomyocytes after CCCP (50 μM) or Thapsi (500 μM) stimulation at 150 s. The results are representative of 4 independent experiments (N = 4), where 10–20 cells were analysed. Data are expressed as mean ± SEM. D) qPCR for MCU mRNA levels of cardiomyocytes transfected with control, MCU (I) or MCU (II) siRNA. E) Western blot of MCU and β-tubulin (upper) and densitometric analysis (lower) of cardiomyocytes transfected with control, MCU (I) or MCU (II) siRNA. Data are expressed as mean ± SEM, N = 3, *P < 0.05, **P < 0.01 and ***P < 0.001 vs. siRNA Control (Ctrl). (PDF 337 KB)
12964_2014_68_MOESM3_ESM.pdf
Additional file 3: Figure S3: Cardiomyocyte hypertrophy induced by NE. A) Representative images of cultured neonatal rat cardiomyocytes treated with or without (Control) NE 10 μM for the indicated times. Cells were fixed and stained with phalloidin-rhodamine (orange) for sarcomere detection and with Höescht (blue) for nuclei detection. Lower panels show the fluorescence intensity profiles of the lines depicted in the respective images. Scale bar: 20 μM. B) Cell area, C) perimeter and D) percentage of sarcomeric cardiomyocytes were calculated measuring at least 100 cells per condition. E) Western blot of β-MHC and β-actin (upper) and densitometric analysis (lower) of cardiomyocytes treated with or without NE 10 μM for the indicated times. Data are expressed as mean ± SEM, N = 3, *P < 0.05, **P < 0.01 and ***P < 0.001 vs Control. (PDF 2 MB)
12964_2014_68_MOESM4_ESM.pdf
Additional file 4: Figure S4.: ER Ca2+ content in hypertrophic cardiomyocytes. A) Cytoplasmic Ca+2 signals induced by thapsigargin 500 μM in control and NE 10 μM or IGF-1 100 nM treated cardiomyocytes. Caffeine 5 mM was added as a negative control. B) Area under the curve of cytoplasmic Ca2+ signals of control and NE and IGF-1 treated cardiomyocytes after thapsigargin 500 μM stimulation between 100 and 300 s. Data are expressed as mean ± SEM, N = 3, **P < 0.01 vs Control. (PDF 312 KB)
12964_2014_68_MOESM5_ESM.pdf
Additional file 5: Figure S5.: IRS-1 phosphorylation in hypertrophic cardiomyocytes. A) Western blot of p-IRS-1 (Ser307) and IRS-1 (left) and densitometric analysis (right) of control and hypertrophic NE treated cardiomyocytes (NE 10 μM, 24 h) either stimulated with insulin 100 nM for 15 min or left unstimulated. Data were relativized against insulin (white bar) and expressed as mean ± SEM, N = 4, ***P < 0.001 vs. insulin. B) Western blot of p-IRS-1 (Ser307) and IRS-1 of control, Ruthenium Red (Ru, 10 μM, 30 min prior stimuli) and xestospongin C (XeC, 100 μM, 30 min prior stimuli) treated cardiomyocytes either stimulated with insulin 100 nM for 15 min or left unstimulated. Data were relativized against insulin (white bar) and expressed as mean ± SEM, N = 3, *P < 0.001 vs. Insulin. (PDF 377 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Gutiérrez, T., Parra, V., Troncoso, R. et al. Alteration in mitochondrial Ca2+ uptake disrupts insulin signaling in hypertrophic cardiomyocytes. Cell Commun Signal 12, 68 (2014). https://doi.org/10.1186/s12964-014-0068-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-014-0068-4