
Abstract
Background
Human gliomas pose significant morbidity and mortality to those afflicted by them, and currently there are no curative treatment modalities available for these highly invasive tumours.
Methods
With the approval from the human ethics committee, patients diagnosed with brain tumour (glioma) were recruited for this study. At the time of surgical resection, freshly resected tumour as well as ‘peri-tumour’ tissue were taken directly from theatre to the laboratory and were successfully cultured. Confocal fluorescence microscopy techniques and immunohistochemistry were used for characterization of human glioma cultures. Dye uptake experiments and confocal microscopy were utilized for P2X7 receptor (P2X7R) pore activity.
Results
We reveal human glioma cultures to contain microglia in close association with glioma (tumour) cells. Both glioma cells and microglia were found to express the purinergic, ATP sensing, P2X7R. P2X7R protein expression was increased in microglia derived from tumour when compared to ‘peri-tumour’ tissue. The pore capacity of P2X7R in tumour-associated microglia was functional, as evidenced by dye uptake experiments. Importantly, inhibition of P2X7R with the synthetic antagonist, brilliant blue G (BBG) resulted in a significant decrease in the number of glioma cells in culture.
Conclusions
P2X7R was found to be over-expressed in grade IV human gliomas and its pore capacity was functional. Antagonism of P2X7R with BBG resulted in a decrease in tumour cell number. This identifies P2X7R as a promising therapeutic target to combat human glioma proliferation.
Similar content being viewed by others

Background
Gliomas are the most common type of intrinsic brain tumour and a major cause of morbidity and mortality for those afflicted by these highly invasive tumours. The majority of adult gliomas are high-grade astrocytomas, comprising grade 3 anaplastic astrocytoma (AA) and grade 4 glioblastoma multiforme (GBM) [1]. In addition to tumour cells, gliomas contain microglia, which are known to contribute to the tumour mass [2]. Microglia are the immunocompetent cells of the central nervous system. Under normal conditions microglia assume a quiescent/resting (ramified) phenotype, but in the setting of brain injury or neoplasia microglia become activated [2]. Activated microglia are capable of releasing various immunomodulatory molecules that could alter the course of tumourogenesis [3]. The mechanisms controlling the transition from ramified to activated microglia are not fully understood. We have recently shown that the purinergic receptor, P2X7R, is involved in this transition [4].
The P2X7R is an ATP sensing receptor expressed in cells of haemopoeitic and immunological origin such as monocytes, macrophages, mast cells and microglia [5]. Increased P2X7R expression in microglia in the brain has been reported in the setting of Alzheimer’s disease [6],[7], multiple sclerosis [8], brain ischemia [9] and spinal cord injury [10]. Pharmacological blockade of P2X7R has been shown to be neuroprotective in an animal model of Alzheimer’s disease [6], enhance recovery in animal models of spinal cord injury [10], and reduce neuroinflammation in an experimental model of autoimmune encephalomyelitis [11]. P2X7R over-expression is also reported in a number of cancers, including those of the breast [12], prostate [12], thyroid [13] pancreas [14], melanoma [15],[16], chronic lymphocytic leukemia [17], human neuroblastoma [18], the rat C6 glioma model, and more recently human glioblastoma [19]. However, the role that P2X7R plays in the biology of brain neoplasms is unknown.
The P2X7R has dual ionic conductance states. Transient stimulation with agonist (most commonly ATP) opens a P2X7R channel permeable to small cations, whereas sustained agonist stimulation leads to a pore state permeable to moieties of up to 900 Da [20],[21]. The P2X7R pore activity has been most commonly associated with consequent cell death, apoptosis or cytolysis [20]-[23]. Recently, we showed that in transfected rat primary hippocampal neuron glia mixed cultures over-expression of P2X7R was sufficient to induce microglial activation and proliferation [4]. The trophic effects observed were dependant on P2X7R pore activity (not channel), and there was no evidence of P2X7R-induced cell death.
Whether P2X7R has a similar action in the setting of human brain tumours is not known. Previous studies have raised questions about the fundamental biological role of P2X7R in the setting of cancer and cell trophism. To explain the over-expression of a purported ‘cytolytic’ receptor in settings of cell trophism, some have suggested that the receptor must therefore be ‘non-functional’ to allow trophism rather than cell death [24]. It has also been argued that in the setting of cancer/cell trophism P2X7R is fully functional (intact channel and pore conductance) and it indeed serves a homeostatic anti-tumour function designed to have pro-apoptotic effects to deal with the growing tumour burden [12]. In contrast some studies have shown no evidence of P2X7R-mediated apoptosis, and attribute the trophism/tumour growth to P2X7R function itself [18].
In this study, we reveal increased P2X7R protein expression in microglia cultured from human brain tumour versus ‘peri-tumour’ (region of macroscopically normal brain surrounding the frank tumour). Pore activity was evident in microglia, indicative of a normally functioning receptor. These observations are also highly supportive of a trophic rather than cell killing role for P2X7R pore. Importantly, total inhibition of P2X7R activity (channel and pore conductances) with brilliant blue G (BBG) reduced the number of tumour cells in culture. The results from this study identify P2X7R as a potential anti-tumour therapeutic target.
Methods
Human tumour and peri-tumour cultures
Protocols for obtaining and handling human brain tissue were reviewed and approved by the Human Research Ethics Committee of the Royal Melbourne Hospital, Victoria, Australia. Written informed consent to study brain tumour/‘peri-tumour’ tissue excised during tumour surgery was obtained from patients prior to the operation. Tumour tissue was obtained during routine tumour resection/debulking, and where safe ‘peri-tumour’ was obtained during the same operation. The ‘peri-tumour’ tissue comprised macroscopically normal brain adjacent to the tumour, which required removal for surgical access. Tissue was received by direct explant from the operating room and placed into sterile containers. Immediately after, the samples were taken to the laboratory and in the laminar flow hood (PC2 laboratory), the tissue was finely chopped. The respective tumour and ‘peri-tumour’ pieces were placed in an enzyme solution containing Papain (200 units; Sigma Aldrich) for 35 minutes at 37°C. The tissue was washed 3 times to remove all traces of papain, and the mixture was triturated to obtain a single cell suspension. The cells were plated into 12 well plates containing 18 mm Poly-D-lysine (Sigma) coated coverslips (SDR Clinical Technology) at a density of 1.8 × 105 cells/well. Cultures were maintained in Minimum Essential Medium (Gibco, Invitrogen) with the following supplements: 1 mm glucose, Penicillin-Streptomycin (5000 units/ml), 10% heat inactivated Fetal Bovine Serum (GIBCO, Invitrogen), MITO + ™ Serum Extender (Becton Dickinson), and 2 mM L-glutamine (GIBCO, Invitrogen). Cells were cultured at 37°C in a humidified incubator of 5% CO2/95% O2. All cultures were from grade IV (histological criteria) gliomas as diagnosed post-hoc after the removal of the tumour.
Immunohistochemistry
Human tumour and ‘peri-tumour’ cultures were fixed in a solution of acetone and methanol (1:1), at −20°C. After fixation, the cells were washed once with phosphate-buffered saline (PBS), and non-specific protein binding sites were blocked with 2% Bovine Serum Albumin (Sigma) for 45 minutes at 37°C. The following primary antibodies were used: rabbit anti-GFAP (glial fibrillary acidic protein) primary antibody (final dilution of 1:400; a kind gift from Professor Jennifer Berka (Department of Immunology, Monash University, Melbourne, Australia), isolectin GS-IB4 from Griffonia simplicifolia, Alexa Fluor® 594 conjugate (final dilution of 1:100; Molecular Probes), goat anti P2X7R antibody (y-14; final dilution 1:100; Quantum Scientific). Primary antibodies were made up in PBS, with 1% Triton-X100 for permeabilization, and were incubated overnight at 4°C. After three, 5 minute washes in PBS, the cells were incubated with the relevant secondary antibodies: Alexa Fluor® 488 (final dilution: 1:200; Molecular Probes) or Texas Red® X (final dilution: 1:200; Molecular Probes). All secondary antibodies were incubated overnight at 4°C. After three, 5 minute washes in PBS, the samples were mounted with DAKO Fluorescent Mounting Medium. No staining was detected in the absence of primary or secondary antibodies. Some preparations were counter labelled with DAPI nuclear stain (5 μM; Molecular Probes).
Confocal microscopy
Human tumour and ‘peri-tumour’ cultures were viewed with a Zeiss LSM 510 META multiphoton/confocal microscope equipped with 488 nm argon, 543 and 633 nm Green and Red Helium/Neon and 800 nm Chameleon lasers. Images were acquired with a 40× IR-Achromat (N.A. 0.80), water immersion objective. For most experiments samples were simultaneously stained with two or three fluorescent probes, with dual or triple emission achieved through appropriately selected emission filters, or by defining emission ranges following prior acquisition of lambda emission profiles for each probe individually. Images were analyzed using MetaMorph (Universal Imaging Corporation®) software for assessment of cell number. For live cell imaging, the cells were bathed in HEPES buffer (mM: NaCl 135, KCl 5, HEPES 10, Glucose 10, CaCl2 1, MgCl2 pH: 7.4) at room temperature (~25°C).
P2X7R pore assay
We measured the degree of P2X7R pore activity inherent in the culture environment in the absence of any exogenous pharmacological stimulus (designated as a pore ‘snap-shot’ experiment). At 7 days post culturing, the cells were exposed to 5 μM of YOPRO-1 (345 kD; Molecular Probes) nuclear dye for 30 minutes. The cultures were then immediately fixed, and mounted for confocal imaging to assess the presence of pores by measuring YOPRO-1 intensity of cell nuclei. To confirm that the response observed was P2X7R specific, some human glioma cultures were pre-exposed to oxidized ATP, a specific and irreversible antagonist of P2X7R [25]. Oxidized ATP (250 μM) was applied 2.5 hours prior to YOPRO-1 application. Thereafter, the cells were washed once in PBS, then immediately fixed, and mounted for confocal imaging to assess the presence of pores by measuring ethidium+ intensity of cell nuclei.
BBG experiments
Prior data from animal studies have revealed that BBG administration protocols that resulted in average tissue concentrations of 9.94-43.59 μM BBG over 3 consecutive days were effective in reducing microglial activation and enhancing recovery after spinal cord injury in rats [26]. We used the more conservative BBG concentration of 7 μM to treat human glioma cultures at day 3 and 5 post culturing. The cultures were left in the presence of the BBG until day 10 post culture. By day 3 tumour and ‘peri-tumour’ cultured cells settled down for adherence to poly-D-lysine coated coverslips, while day 5 was routinely chosen as the day to replenish the culture medium. Thereafter, the cultures were fixed with a solution of acetone:methanol (50:50), and processed for immunohistochemistry as described above.
Results
Morphological and cellular characteristics of human gliomas in culture
We were able to successfully grow human tumour (n = 10 patients) and ‘peri-tumour’ cultures (n = 10 patients). Figure 1 shows human glioma cultures after 7 days of culturing, and illustrates extensive arrays of tumour cells (GFAP positive) dispersed throughout the culture environment. Tumour cells were found to cluster/aggregate forming a densely packed arrangement (Figure 1A,C). Within each cluster there were highly pleomorphic cells with multiple nuclei (Figure 1A). This aggregative capacity was absent from cultures derived from ‘peri-tumour’ tissue (n = 4) peri-tumour cultures).

Human glioma cultures. A: Cell clusters (arrow) noted in glioma cultures. GFAP (red) and DAPI (blue). Arrowhead showing evidence of nuclear atypia. Scale 100 μM. B: Glioma cultures labeled with GFAP (Red) showing the highly heterogeneous morphology of these cells. Scale 20 μm. C: Microglia also formed clusters in glioma cultures. Red = microglial marker, isolectin GS-IB4. Scale 50 μm.
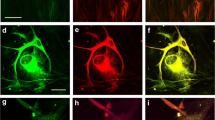
Microglia infiltrated the clusters of tumour cells, with microglia and tumour cells being in very close proximity to one another (Figure 2). Whilst GFAP-positive tumour cells were concentrated in the periphery of the cell clusters, microglia were found at the core of the cluster (Figure 2).

The cell clusters evident in human glioma cultures consisted of glioma cells and microglia. (A to K) Consecutive z-slices of the cell cluster, each image (slice) taken at 15 μm from the top of the cluster to the bottom. Microglia (isolectin GS-IB4 positive, red) formed the core of the cluster surrounded by glioma cells (GFAP positive, green). Scale, 50 μm.
P2X7R is over-expressed in microglia found in human glioma cultures
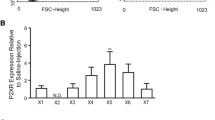
Cell in these clusters were also found to express P2X7R (Figure 3). The expression of P2X7R was noted in both tumour cells (Figure 3A,B) and microglia (Figure 3C,D). Microglia derived from tumour tissue showed higher levels of P2X7R protein expression than microglia derived from ‘peri-tumour’ tissue (Figure 4A). Conversely, tumour cells derived from the core of the glioma were found to have lower P2X7R expression than tumour cells from ‘peri-tumour’ tissue (Figure 4B).

The human glioma cell clusters in culture were P2X7R positive. A: Cell cluster of glioma cells (GFAP positive, red) also expressed P2X7R receptor (green). Scale, 100 μm. B: Magnified image of a glioma cell (GFAP positive, red), also expressing P2X7R (green). Image in the right showing the co-localization of GFAP and immunoreactivity for P2X7R. Scale, 50 μm. C: Cell cluster in human glioma culture labeled for microglia (isolectin GS-IB4, red), also showed P2X7R immunoreactivity (green). Scale, 50 μm. D: Magnified image of a microglia (isolectin GS-IB4 positive, red) showing expression of P2X7R (green). Right image shows co-localization of isolectin GS-IB4 and P2X7R immunoreactivity. Scale, 10 μm.

Expression of P2X7R in tumour versus ‘peri- tumour’. P2X7R expression was measured using immunohistochemistry against P2X7R protein. The degree of fluorescence intensity (standardized to background fluorescence intensity) in each cell was used as an indication of P2X7R expression. A. P2X7R expression was increased in microglia derived from tumour versus ‘peri-tumour’ tissue. n = 517 cells from 4 human glioma (tumour) and respective ‘peri-tumour’ tissue. Insets above the graph show representative images of microglia expressing P2X7R. Red = microglial marker isolectin GS-IB4. Green = P2X7R expression. Scale bar, 20 μm. B. P2X7R expression was conversely increased in ‘peri-tumour’ glioma cells compared to glioma cells derived from the core of the tumour. n = 551 cells from 3 human glioma tumour and respective ‘peri-tumour’ tissue. Insets above the graph show representative images of glioma cells expressing P2X7R. Red = glioma cell marker, GFAP, Green = P2X7R expression. Scale bar, 20 μm.
The pore capacity of P2X7R is active in human glioma associated microglia
The incorporation of YOPRO-1, a high molecular weight fluorescence dye, indicates the presence of active P2X7R pores in human glioma microglia (Figure 5). YOPRO-1 incorporation was inhibited by blocking P2X7R activity with a specific and irreversible antagonist, oxATP, confirming the response was P2X7R specific (Figure 5). A significantly higher level of YOPRO-1 was incorporated into the nuclei of the glioma microglia suggesting that pore activity may be having a similar pro-activation (trophic) role to that previously described in rat hippocampal microglia [4],[27].

Microglia from human glioma at 3 days post culturing showed P2X7R pore activity, as evidenced by dye uptake experiments. YOPRO-1 nuclear intensity was measured as an index of P2X7R pore capacity (n = 42 microglia being isolectin GS-IB4 positive from 3 human gliomas). This pore capacity was reduced with inhibition with a specific and irreversible antagonist of P2X7R, oxATP (250 μM of oxATP for 3 hours prior to YOPRO-1 incubation, n = 28 microglia, being isolectin GS-IB4 positive from 3 human gliomas).
Inhibition of P2X7R activity was found to lead to a decrease in the number of tumour cells
When glioma cultures were incubated with BBG (an antagonist of P2X7R) after 3 and 5 days in culture, there were significantly fewer microglia and a reduced number of glioma cells in culture by day 10 (Figure 6).

Inhibition of P2X7R with BBG reduced the number of glioma cells and microglia in human glioma cultures. Representative human glioma cell cultures before (A. control) and after (B. BBG) treatment with brilliant blue G. Red = isolectin GS IB4. Blue = DAPI nuclear stain. Scale bar, 20 μm. C. Treatment of human glioma cultures with 7 μM BBG at day 3 and day 5 post culture reduced the number of microglial cells. The number of microglia were counted using immunohistochemistry against a microglial marker, isolectin GS IB4 at 10 days after culturing. Nuclei were counter labeled with DAPI nuclear stain. Cell numbers were counted using MetaMorph imaging software. n = 109 randomly selected fields from 3 human gliomas that were not treated with BBG (control). n = 107 randomly selected fields from 3 human gliomas that were treated with BBG (BBG treated). D. Treatment of human glioma cultures with 7 μM BBG at day 3 and day 5 post culture reduced the number gliomas cells. The number of glioma cells were counted using immunohistochemistry against GFAP at 10 days after culturing. Nuclei were counter labeled with DAPI nuclear stain. Cell numbers were counted using MetaMorph imaging software. n = 114 randomly selected fields from 7 human gliomas that were not treated with BBG (control). n = 113 randomly selected fields from 7 human gliomas that were treated with BBG (BBG treated).
Discussion
Human brain tumours grown in culture comprise microglia that are dispersed throughout and within the core of the tumour cells. Here we showed that both microglia and tumour cells expressed P2X7R, with higher expression levels in microglia associated with the tumour when compared to the ‘peri-tumour’. Importantly, P2X7R pore activity was evident in tumour-associated microglia. Inhibition of overall P2X7R function led to a decrease in tumour cell number, revealing P2X7R as a potential anti-tumour therapeutic candidate.
In the culture environment, human brain tumours were found to have a significant number of microglia in close association with the tumour cells. Microglia within astrocytomas are known to proliferate along the architecture formed by tumour cells [28]. In addition, astrocytoma cells produce the microglia and monocyte chemoattractant proten-1 (MCP-1) [29] with microglia expressing the MCP-1 receptor, CCR2 [30]. Hence, increased presence of microglia at the site of astrocytic glioma may result from recruitment of microglia, as well as local microglial proliferation. Interestingly, tumours with a higher proliferation rate such as GBM contain significantly higher numbers of activated microglia compared to tumours of lower grade [2]. Similarly, a positive correlation has been found between the number of CD11b-positive microglia/macrophages in gliomas and the proliferative capacity of the tumour as indexed by bromodeoxyuridine (BrdU)+ or Ki-67+ labelling [31]. The proliferative capacity of microglia within human brain tumours should therefore be taken into account when histologically characterizing tumour progression/invasion and potentially determining patient prognosis.
Interestingly, we found P2X7R expression to be enhanced in ‘peri-tumour’ glioma cells compared to tumour-associated glioma cells, suggesting that this may play a role actively invading glioma cells. Conversely, P2X7R expression was increased in tumour-associated microglia compared to microglia derived from the ‘peri-tumour’ brain. P2X7R up-regulation has been shown in many different cancer types, but the majority of these studies have not defined the locality (i.e., tumour or ‘peri-tumour’ region) of that expression. Whether P2X7R functions as a pro-apoptotic/cytolytic entity or conversely leads to tumour growth is also not fully understood. There are observations of enhanced P2X7R expression in solid epithelial tumours [12] and strong up-regulation of P2X7R in samples from thyroid cancer lines [32]. P2X7R up-regulation was also shown in thyroid papillary cancer, and the receptor was found to be fully functional [32], the authors suggesting P2X7R as a possible biomarker of the disease. In prostate cancer P2X7R expression was increased [33], and a monoclonal anti-P2X7R antibody is currently being designed for use as a prostate cancer biomarker. Similarly, in newly diagnosed pediatric acute leukemias P2X7R expression was increased, especially in cases of relapsed disease and the receptor was found to be fully functional [34]. In rat C6 glioma cells P2X7R mRNA and protein were up-regulated upon exposure to P2X7R specific agonist BzATP, and the stimulation of P2X7R was linked to release of proinflammatory markers and tumour cell migration [35]. P2X7R stimulation with ATP was found to be important in maintenance of survival of Neuro-2a neuroblastoma cells [36]. In neuroblastoma cells, stimulation of P2X7R resulted in cell shrinkage and plasma membrane blebbing, with no signs of apoptosis or necrosis, but with evidence of proliferation [18]. Similarly, immunohistochemical data revealed increased P2X7R expression in Schwann cells from human lingual nerve neuromas [37] and in human gliomas [19]. In this latter study P2X7R expression in glioma tissue was compared with brain tissue from individuals with other neurological conditions. No distinction could be made between tumour and ‘peri-tumour’ tissue within the same individual and the specific cell contributions (microglia versus glioma cells) in the increased P2X7R expression was also not defined [19].
So while we have shown that microglia occur in close association to human brain tumour cells and that they have increased P2X7R expression, do microglia and P2X7R actually contribute to tumour growth/proliferation? In primary hippocampal cultures we have previously shown that P2X7R pore activity (not channel) drives microglial activation and proliferation as well as inducing the release of inflammatory mediators [4]. This finding was somewhat surprising given that this conductance state of the P2X7R was widely held to be cytolytic [20],[21],[24],[38]-[40] rather than trophic. Similarly, in B-cell chronic lymphocytic leukemia where P2X7R was shown to be over-expressed, the authors suggested that the receptor must be non-functional producing an anti-apoptotic effect and leading to accumulation of tumour cells [24]. In C6 glioma cell line P2X7R suppression was also shown to cause cell proliferation with angiogenesis, effects that were mediated by epidermal growth factor receptor [41]. Again, in this study no distinction was made between P2X7R pore versus channel activity. In contrast, we found glioma-associated microglia to have P2X7R pore, which was functional. Unlike the current dogma, it is possible that P2X7R pore has a trophic rather than apoptotic role in gliomas.
The current treatment options available for human high-grade astrocytoma include maximal safe surgical resection followed by radiotherapy with or without chemotherapy. However, although aggressive management can extend life, there is no cure and there remains a prognostic period of approximately six to twelve months especially for high-grade tumours [42]. The addition of targeted agents has received intense interest recently. The role of chemotherapy or radiotherapy alone remains limited [43],[44]. Hence, more effective treatment modalities are needed. BBG is an analogue of brilliant blue FCF (FD&C blue dye No. 1) which is a synthetic dye approved by the Food and Drug Administration as a food additive considered as one of the safest dyes currently available, with no toxicity at doses of up to 12 mg/kg per day in healthy animals [45]. BBG is also a commonly used synthetic antagonist of P2X7R [46] with the capacity to penetrate the blood brain barrier [26]. In an animal model, average tissue concentrations of 9.94 - 43.49 μM BBG administered reduced microglial activation and improved recovery after spinal cord injury [26]. No adverse effects on behaviour, weight, survival, or other physiological parameters, including body temperature, blood pH, blood gases, or blood pressure were observed. On this basis, we used even a lower (presumably ‘safer’) dose of BBG (7 μM) that was found to be effective in decreasing the number of microglia and glioma cells. It is possible that BBG acts either directly on P2X7Rs expressed on tumour cells, or indirectly via inhibiting P2X7Rs on tumour-associated microglia. We have shown over-expression of P2X7R alone to be sufficient in inducing microglial activation and proliferation [4]. Once activated, microglia are known to release factors such as vascular endothelial growth factor (VEGF) [47] and other cytokines (i.e., including IL-6 and TNF-α) involved in angiogenesis and tumour cell migration [48]-[50]. By driving microglial activation and proliferation with concomitant release of bioactive tumorigenic factors, it can be postulated that P2X7R plays a key role in the molecular hierarchy of glioma development.
Conclusions
In conclusion, this study reveals human glioma cultures to show large numbers of microglia in close association with tumour cells. Both microglia and the tumour cells were found to express P2X7R whose expression was greater in tumour associated microglia versus ‘peri-tumour’ tissue. Importantly, P2X7R pore was active in these microglia, and inhibition of P2X7R function resulted in a decrease in the number of glioma cells. Overall, our data support P2X7R antagonism as a potential therapeutic avenue for treatment of human gliomas.
Abbreviations
- AA:
-
Anaplastic astrocytoma
- GBM:
-
Gliomblastoma multiforme
- P2X7R:
-
P2X7 receptor
- BBG:
-
Brilliant blue G
- GFAP:
-
Glial fibrillary acidic protein
- VEGF:
-
Vascular endothelial growth factor
- MCP-1:
-
Monocyte chemoattractant proten-1
- TNF-α:
-
Tumour necrosis factor alpha
- IL-6:
-
Interleukin 6
- YOPRO:
-
Carbocyanine nucleic acid stain
References
Kleihues P, Burger PC, Scheithauer BW: The new WHO classification of brain tumours. Brain Pathol. 1993, 3: 255-268. 10.1111/j.1750-3639.1993.tb00752.x.
Graeber MB, Scheithauer BW, Kreutzberg GW: Microglia in brain tumors. Glia. 2002, 40: 252-259. 10.1002/glia.10147.
Wesolowska A, Kwiatkowska A, Slomnicki L, Dembinski M, Master A, Sliwa M, Franciszkiewicz K, Chouaib S, Kaminska B: Microglia-derived TGF-beta as an important regulator of glioblastoma invasion–an inhibition of TGF-beta-dependent effects by shRNA against human TGF-beta type II receptor. Oncogene. 2008, 27: 918-930. 10.1038/sj.onc.1210683.
Monif M, Reid CA, Powell KL, Smart ML, Williams DA: The P2X7 receptor drives microglial activation and proliferation: a trophic role for P2X7R pore. J Neurosci. 2009, 29: 3781-3791. 10.1523/JNEUROSCI.5512-08.2009.
Ralevic V, Burnstock G: Receptors for purines and pyrimidines. Pharmacol Rev. 1998, 50: 413-492.
Parvathenani LK, Tertyshnikova S, Greco CR, Roberts SB, Robertson B, Posmantur R: P2X7 mediates superoxide production in primary microglia and is up-regulated in a transgenic mouse model of Alzheimer’s disease. J Biol Chem. 2003, 278: 13309-13317. 10.1074/jbc.M209478200.
Rye JA, O’Hara Tompkins N, Eck R, Neal WA: Promoting youth physical activity and healthy weight through schools. W V Med J. 2008, 104: 12-15.
Yiangou Y, Facer P, Durrenberger P, Chessell IP, Naylor A, Bountra C, Banati RR, Anand P: COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 2006, 6: 12-10.1186/1471-2377-6-12.
Franke H, Gunther A, Grosche J, Schmidt R, Rossner S, Reinhardt R, Faber-Zuschratter H, Schneider D, Illes P: P2X7 receptor expression after ischemia in the cerebral cortex of rats. J Neuropathol Exp Neurol. 2004, 63: 686-699.
Wang X, Arcuino G, Takano T, Lin J, Peng WG, Wan P, Li P, Xu Q, Liu QS, Goldman SA, Nedergaard M: P2X7 receptor inhibition improves recovery after spinal cord injury. Nat Med. 2004, 10: 821-827. 10.1038/nm1082.
Matute C, Torre I, Perez-Cerda F, Pérez-Samartín A, Alberdi E, Etxebarria E, Arranz AM, Ravid R, Rodríguez-Antigüedad A, Sánchez-Gómez M, Domercq M: P2X(7) receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J Neurosci. 2007, 27: 9525-9533. 10.1523/JNEUROSCI.0579-07.2007.
Slater M, Danieletto S, Pooley M, Cheng Teh L, Gidley-Baird A, Barden JA: Differentiation between cancerous and normal hyperplastic lobules in breast lesions. Breast Cancer Res Treat. 2004, 83: 1-10. 10.1023/B:BREA.0000010670.85915.0f.
Pines A, Bivi N, Vascotto C, Romanello M, D'Ambrosio C, Scaloni A, Damante G, Morisi R, Filetti S, Ferretti E, Quadrifoglio F, Tell G: Nucleotide receptors stimulation by extracellular ATP controls Hsp90 expression through APE1/Ref-1 in thyroid cancer cells: a novel tumorigenic pathway. J Cell Physiol. 2006, 209: 44-55. 10.1002/jcp.20704.
Kunzli BM, Berberat PO, Giese T, Csizmadia E, Kaczmarek E, Baker C, Halaceli I, Büchler MW, Friess H, Robson SC: Upregulation of CD39/NTPDases and P2 receptors in human pancreatic disease. Am J Physiol Gastrointest Liver Physiol. 2007, 292: G223-G230. 10.1152/ajpgi.00259.2006.
Deli T, Varga N, Adam A, Kenessey I, Rásó E, Pusk’s LG, Tóvári J, Fodor J, Fehér M, Szigeti GP, Csernoch L, Tímár J: Functional genomics of calcium channels in human melanoma cells. Int J Cancer. 2007, 121: 55-65. 10.1002/ijc.22621.
White N, Butler PE, Burnstock G: Human melanomas express functional P2 X(7) receptors. Cell Tissue Res. 2005, 321: 411-418. 10.1007/s00441-005-1149-x.
Adinolfi E, Melchiorri L, Falzoni S, Chiozzi P, Morelli A, Tieghi A, Cuneo A, Castoldi G, Di Virgilio F, Baricordi OR: P2X7 receptor expression in evolutive and indolent forms of chronic B lymphocytic leukemia. Blood. 2002, 99: 706-708. 10.1182/blood.V99.2.706.
Raffaghello L, Chiozzi P, Falzoni S, Di Virgilio F, Pistoia V: The P2X7 receptor sustains the growth of human neuroblastoma cells through a substance P-dependent mechanism. Cancer Res. 2006, 66: 907-914. 10.1158/0008-5472.CAN-05-3185.
Ryu JK, Jantaratnotai N, Serrano-Perez MC, McGeer PL, McLarnon JG: Block of Purinergic P2X7R Inhibits Tumor Growth in a C6 Glioma Brain Tumor Animal Model. J Neuropathol Exp Neurol. 2011, 70: 13-22. 10.1097/NEN.0b013e318201d4d4.
Di Virgilio F: The P2Z purinoceptor: an intriguing role in immunity, inflammation and cell death. Immunol Today. 1995, 16: 524-528. 10.1016/0167-5699(95)80045-X.
Surprenant A, Rassendren F, Kawashima E, North RA, Buell G: The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science. 1996, 272: 735-738. 10.1126/science.272.5262.735.
Schilling WP, Wasylyna T, Dubyak GR, Humphreys BD, Sinkins WG: Maitotoxin and P2Z/P2X(7) purinergic receptor stimulation activate a common cytolytic pore. Am J Physiol. 1999, 277: C766-C776.
Wang Q, Wang L, Feng YH, Li X, Zeng R, Gorodeski GI: P2X7 receptor-mediated apoptosis of human cervical epithelial cells. Am J Physiol Cell Physiol. 2004, 287: C1349-C1358. 10.1152/ajpcell.00256.2004.
Wiley JS, Dao-Ung LP, Gu BJ, Sluyter R, Shemon AN, Li C, Taper J, Gallo J, Manoharan A: A loss-of-function polymorphic mutation in the cytolytic P2X7 receptor gene and chronic lymphocytic leukaemia: a molecular study. Lancet. 2002, 359: 1114-1119. 10.1016/S0140-6736(02)08156-4.
Murgia M, Hanau S, Pizzo P, Rippa M, Di Virgilio F, Oxidized ATP: An irreversible inhibitor of the macrophage purinergic P2Z receptor. J Biol Chem. 1993, 268: 8199-8203.
Peng W, Cotrina ML, Han X, Yu H, Bekar L, Blum L, Takano T, Tian GF, Goldman SA, Nedergaard M: Systemic administration of an antagonist of the ATP-sensitive receptor P2X7 improves recovery after spinal cord injury. Proc Natl Acad Sci U S A. 2009, 106: 12489-12493. 10.1073/pnas.0902531106.
Monif M, Burnstock G, Williams DA: Microglia: proliferation and activation driven by the P2X7 receptor. Int J Biochem Cell Biol. 2010, 42: 1753-1756. 10.1016/j.biocel.2010.06.021.
Klein R, Roggendorf W: Increased microglia proliferation separates pilocytic astrocytomas from diffuse astrocytomas: a double labeling study. Acta Neuropathol. 2001, 101: 245-248.
Prat E, Baron P, Meda L, Scarpini E, Galimberti D, Ardolino G, Catania A, Scarlato G: The human astrocytoma cell line U373MG produces monocyte chemotactic protein (MCP)-1 upon stimulation with beta-amyloid protein. Neurosci Lett. 2000, 283: 177-180. 10.1016/S0304-3940(00)00966-6.
Galasso JM, Stegman LD, Blaivas M, Harrison JK, Ross BD, Silverstein FS: Experimental gliosarcoma induces chemokine receptor expression in rat brain. Exp Neurol. 2000, 161: 85-95. 10.1006/exnr.1999.7249.
Morimura T, Neuchrist C, Kitz K, Budka H, Scheiner O, Kraft D, Lassmann H: Monocyte subpopulations in human gliomas: expression of Fc and complement receptors and correlation with tumor proliferation. Acta Neuropathol. 1990, 80: 287-294. 10.1007/BF00294647.
Solini A, Cuccato S, Ferrari D, Santini E, Gulinelli S, Callegari MG, Dardano A, Faviana P, Madec S, Di Virgilio F, Monzani F: Increased P2X7 receptor expression and function in thyroid papillary cancer: a new potential marker of the disease?. Endocrinology. 2008, 149: 389-396. 10.1210/en.2007-1223.
Slater M, Danieletto S, Gidley-Baird A, Teh LC, Barden JA: Early prostate cancer detected using expression of non-functional cytolytic P2X7 receptors. Histopathology. 2004, 44: 206-215. 10.1111/j.0309-0167.2004.01798.x.
Chong JH, Zheng GG, Zhu XF, Guo Y, Wang L, Ma CH, Liu SY, Xu LL, Lin YM, Wu KF: Abnormal expression of P2X family receptors in Chinese pediatric acute leukemias. Biochem Biophys Res Commun. 2010, 391: 498-504. 10.1016/j.bbrc.2009.11.087.
Wei W, Ryu JK, Choi HB, McLarnon JG: Expression and function of the P2X(7) receptor in rat C6 glioma cells. Cancer Lett. 2008, 260: 79-87. 10.1016/j.canlet.2007.10.025.
Wu PY, Lin YC, Chang CL, Lu HT, Chin CH, Hsu TT, Chu D, Sun SH: Functional decreases in P2X7 receptors are associated with retinoic acid-induced neuronal differentiation of Neuro-2a neuroblastoma cells. Cell Signal. 2009, 21: 881-891. 10.1016/j.cellsig.2009.01.036.
Morgan CR, Bird EV, Robinson PP, Boissonade FM: Immunohistochemical analysis of the purinoceptor P2X7 in human lingual nerve neuromas. J Orofac Pain. 2009, 23: 65-72.
Nuttle LC, Dubyak GR: Differential activation of cation channels and non-selective pores by macrophage P2z purinergic receptors expressed in Xenopus oocytes. J Biol Chem. 1994, 269: 13988-13996.
Pizzo P, Murgia M, Zambon A, Zanovello P, Bronte V, Pietrobon D, Di Virgilio F: Role of P2z purinergic receptors in ATP-mediated killing of tumor necrosis factor (TNF)-sensitive and TNF-resistant L929 fibroblasts. J Immunol. 1992, 149: 3372-3378.
Rassendren F, Buell G, Newbolt A, North RA, Surprenant A: Identification of amino acid residues contributing to the pore of a P2X receptor. Embo J. 1997, 16: 3446-3454. 10.1093/emboj/16.12.3446.
Fang J, Chen X, Zhang L, Chen J, Liang Y, Li X, Xiang J, Wang L, Guo G, Zhang B, Zhang W: P2X7R suppression promotes glioma growth through epidermal growth factor receptor signal pathway. Int J Bioche Cell Biol. 2013, 45 (6): 1109-1120. 10.1016/j.biocel.2013.03.005.
Hayes RL, Koslow M, Hiesiger EM, Hymes KB, Hochster HS, Moore EJ, Pierz DM, Chen DK, Budzilovich GN, Ransohoff J: Improved long term survival after intracavitary interleukin-2 and lymphokine-activated killer cells for adults with recurrent malignant glioma. Cancer. 1995, 76: 840-852. 10.1002/1097-0142(19950901)76:5<840::AID-CNCR2820760519>3.0.CO;2-R.
Cher L, Rosenthal MA, Drummond KJ, Dally M, Murphy M, Ashley D, Thursfield V, Giles GG: The use of chemotherapy in patients with gliomas: patterns of care in Victoria from 1998-2000. J Clin Neurosci. 2008, 15: 398-401. 10.1016/j.jocn.2007.04.001.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO: Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005, 352: 987-996. 10.1056/NEJMoa043330.
Borzelleca JF, Depukat K, Hallagan JB: Lifetime toxicity/carcinogenicity studies of FD & C Blue No. 1 (brilliant blue FCF) in rats and mice. Food Chem Toxicol. 1990, 28: 221-234. 10.1016/0278-6915(90)90034-K.
Jiang LH, Mackenzie AB, North RA, Surprenant A: Brilliant blue G selectively blocks ATP-gated rat P2X(7) receptors. Mol Pharmacol. 2000, 58: 82-88.
Lafuente JV, Adan B, Alkiza K, Garibi JM, Rossi M, Cruz-Sanchez FF: Expression of vascular endothelial growth factor (VEGF) and platelet-derived growth factor receptor-beta (PDGFR-beta) in human gliomas. J Mol Neurosci. 1999, 13: 177-185. 10.1385/JMN:13:1-2:177.
Palma C, Urbani F, Manzini S: Interleukin-6 production by U373 MG, a human astrocytoma cell line: different pathways involved in substance P and lipopolysaccharide activation. J Neuroimmunol. 1995, 59: 155-163. 10.1016/0165-5728(95)00040-9.
Luber-Narod J, Kage R, Leeman SE: Substance P enhances the secretion of tumor necrosis factor-alpha from neuroglial cells stimulated with lipopolysaccharide. J Immunol. 1994, 152: 819-824.
Sliwa M, Markovic D, Gabrusiewicz K, Synowitz M, Glass R, Zawadzka M, Wesolowska A, Kettenmann H, Kaminska B: The invasion promoting effect of microglia on glioblastoma cells is inhibited by cyclosporin A. Brain. 2007, 130: 476-489. 10.1093/brain/awl263.
Acknowledgements
This study was supported in part by an NHMRC Project Grant (#628301) to TOB and KD and an ARC Discovery Grant (DP0770955) to DAW.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MM: conception and design, collection of tumours from theatre, culturing of tumours, immunohistochemistry, confocal microscopy, dye up-take experiments, results analysis, and drafting manuscript. TJO: conception and design of the study and interpretation of data. CAR: conception and design of experiments, interpretation of data, and editing of manuscript. KJD: neurosurgeon, acquisition of samples, conception and design of experiments. SVL: patient recruitment and consent. DAW: conception and design of experiments, analysis, interpretation of data, assisted with drafting, graphics and editing manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Monif, M., O’Brien, T.J., Drummond, K.J. et al. P2X7 receptors are a potential novel target for anti-glioma therapies. J Inflamm 11, 25 (2014). https://doi.org/10.1186/s12950-014-0025-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12950-014-0025-4