
Abstract
Background
Potentially lethal and heritable cardiomyopathies and cardiac channelopathies are caused by heterogeneous autosomal dominant mutations in over 50 distinct genes, and multiple genes are responsible for a given disease. Clinical genetic tests are available for several of the inherited cardiac diseases and clinical investigations guide which test to order. This study describes a family with cardiac disease in which marked clinical diversity exists. In the absence of a unified clinical diagnosis, we used exome sequencing to identify a causal mutation.
Methods
Clinical evaluation of family members was performed, including physical examination, electrocardiography, 2D transthoracic echocardiography and review of autopsy records. Exome sequencing was performed on a clinically affected individual and co-segregation studies and haplotype analysis were performed to further confirm pathogenicity.
Results
Clinically affected members showed marked cardiac phenotype heterogeneity. While some individuals were asymptomatic, other presentations included left ventricular non-compaction, a resuscitated cardiac arrest due to idiopathic ventricular fibrillation, dilated cardiomyopathy, and sudden unexplained death. Whole exome sequencing identified an Ala119Thr mutation in the alpha-actinin-2 (ACTN2) gene that segregated with disease. Haplotype analysis showed that this mutation segregated with an identical haplotype in a second, previously described family with clinically diverse cardiac disease, and is likely inherited from a common ancestor.
Conclusions
Mutations in the ACTN2 gene can be responsible for marked cardiac phenotype heterogeneity in families. The diverse mechanistic roles of ACTN2 in the cardiac Z-disc may explain this heterogeneous clinical presentation. Exome sequencing is a useful adjunct to cardiac genetic testing in families with mixed clinical presentations.
Similar content being viewed by others

Background
Concerted effort over the past two decades has identified over 50 distinct genes that cause potentially lethal and heritable cardiomyopathies and primary arrhythmogenic disorders. For most of these disorders, there are multiple disease-associated genes [1],[2]. Furthermore, mutations in a single gene can cause different diseases, e.g. mutations in the myosin heavy chain 7 (MYH7) gene cause hypertrophic cardiomyopathy (HCM), restrictive cardiomyopathy and dilated cardiomyopathy (DCM) [3]-[5], while mutations in the sodium channel, type V, alpha subunit (SCN5A) gene cause long QT syndrome (LQTS) and Brugada syndrome [6],[7]. Even within families harbouring the same mutation, incomplete penetrance, phenotype heterogeneity and a variable risk of adverse outcomes are common. In spite of the low genotype-phenotype correlation in the cardiomyopathies and cardiac channelopathies, recent guidelines and expert opinion recommend genetic testing on index cases with a sound clinical suspicion for disease [8], as a genetic diagnosis can explain why the disease has occurred, and allows predictive testing of other asymptomatic at-risk family members.
Commercial genetic tests are available for several of the inherited cardiac diseases, and clinical evaluations guide which test to order. Typically, only the genes most frequently associated with a given disease are screened for mutations. However, advances in targeted massively parallel sequencing, or next-generation sequencing, have overcome the limitations of traditional direct DNA sequencing, and ever-expanding panels of genes are available for screening, including all of the protein-coding genes, i.e. the exome [9],[10],
This study describes the clinical and genetic investigations of an Australian family with marked cardiac phenotype heterogeneity among four individuals, including left ventricular non-compaction (LVNC), idiopathic ventricular fibrillation, DCM and sudden unexplained death, which lent itself to the application of exome sequencing to identify a pathogenic mutation.
Methods
Clinical evaluation of family members
Clinical evaluation of family members was performed at the Genetic Heart Diseases Clinic at Royal Prince Alfred Hospital, Sydney, and Melbourne Heart Centre, The Royal Melbourne Hospital, Parkville, Australia, with written informed consent and in accordance with the local human ethics standards. Clinical evaluation included detailed personal and family histories, physical examination, transthoracic echocardiography, 12-lead electrocardiogram (ECG) recording, cardiac magnetic resonance imaging (MRI), 24-hour Holter monitoring, exercise testing and review of medical records and autopsy reports.
Exome sequencing
Genomic DNA was extracted from buffy coat using a QIAmp DNA blood mini kit (Qiagen, Limburg, NL), according to manufacturer’s recommendations. An adaptor-ligated DNA sequencing library was prepared, enriched for the Illumina TruSeq Exome, and paired-end sequenced using an Illumina HiSeq2000 (Macrogen Facility, Seoul, Korea). The 101 bp sequences were aligned to the human genome sequence (GRCh37/hg19) using BWA v0.7.4 [11] with the default parameters. Sequence alignment files were sorted, converted to binary format and indexed using SamTools v0.1.19 [12], and duplicate reads were removed using Picard tools v1.81 (http://picard.sourceforge.net/index.shtml). The Genome Analysis Tool Kit v2.7.2 [13] (GATK) was used for read realignment around insertions/deletions, base quality score recalibration and genotyping of simple nucleotide variations (SNVs) and short insertions and deletions (InDels) using UnifiedGenotyper, according to the GATK best practices (http://www.broadinstitute.org/gatk/guide/best-practices). SNVs and InDels were annotated using SeattleSeq Annotation tool v8.07 (http://snp.gs.washington.edu/SeattleSeqAnnotation137/index.jsp) and compared against the November 2010 release of the 1000 Genomes Project data (http://www.1000genomes.org/), the National Heart, Lung and Blood Institute Exome Sequencing Project (ESP) data (http://evs.gs.washington.edu/evs_bulk_data/ESP6500SI-V2-SSA137.GRCh38-liftover.snps_indels.txt.tar.gz) and in-house exome sequences of 96 unrelated individuals using custom Perl scripts. Estimates of gene expression levels (FKPM) in cardiac tissue were determined with a custom built RNASeq analysis pipeline using data from Illumina’s Human BodyMap 2.0 project (ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR030/ERR030886/ERR030886_1.fastq.gz; ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR030/ERR030886/ERR030886_2.fastq.gz).
Variant validation using Sanger sequencing
For validation of detected variants, genomic regions up to 500 bp surrounding variants of interest were PCR amplified, excess primers and deoxynucleotide triphosphates removed using alkaline phosphatase (New England Biolabs, MA, USA) and exonuclease I (New England Biolabs), respectively, and Sanger DNA sequenced (Macrogen). Sequencing electropherograms were manually inspected using Sequencher v5.1 (Gene Codes Corp, MI, USA).
Haplotype analysis
Primer sequences were designed, with a 6-FAM fluorophore on the reverse primer, to PCR amplify a variable number of tandem repeat (VNTR) polymorphism in alpha-actinin 2 (ACTN2), and ryanodine receptor 2 (RYR2), plus markers D1S2670, D1S285 and D1S2678 (Additional file 1). PCR was performed at 95°C for 2 minutes, followed by 30 cycles of 95°C for 30 seconds, 54°C for 30 seconds and 72°C for 30 seconds, followed by a final extension at 72°C for 5 minutes. PCR amplicons were separated on an ABI 3730xl DNA analyser (Macrogen) with GeneScan™ 500 LIZ™ size standard (Life Technologies, CA, USA) and alleles sized using Peak ScannerTM software v1.0 (Life Technologies).
All studies were conducted with strict approval and in accordance with the Sydney Local Health District Ethics Review Committee (RPAH zone), Australia.
Results
Clinical characterisation of family ALB
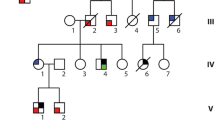
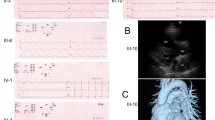
The clinical characteristics of family ALB are summarised in Table 1 and the pedigree illustrated in Figure 1. The family demonstrates marked clinical heterogeneity. The female proband (III:5) presented at age 22 years for clinical screening with a history of syncope and a family history of premature sudden unexplained death. Her sister (III:6) died suddenly during sleep at age 25 years, and her post-mortem failed to identify a cause of death. The proband’s ECG showed sinus rhythm with non-specific ST-T wave changes, whilst her echocardiogram and MRI showed prominent left ventricular apical trabeculations with preserved left ventricular systolic function, consistent with LVNC (Figure 2). There were no inducible arrhythmias during an electrophysiological study, and her QTc measured 440 ms. An implantable cardioverter defibrillator (ICD) was subsequently implanted and she remains stable, with no ICD discharges to date.

ALB Family Pedigree. Squares - males; Circles - females; Line through symbol – deceased individual; open symbol with N – clinically unaffected individual; LVNC - left ventricular non-compaction; VF – ventricular fibrillation; DCM – dilated cardiomyopathy; SUD – sudden unexplained death.

ECG and echocardiographic features of the proband (III:5). The ECG (A) shows minimal pathological changes, while the echocardiogram shows (B) LVNC predominantly affecting the left ventricular apex (arrowed) and (C) colour Doppler flow between the trabeculations.
The proband’s father (II:3) had a history of dyspnoea, left bundle branch block and left ventricular dilatation, with a reduced left ventricular ejection fraction of 27%. The cause of his DCM remained unclear. One female cousin of the proband (III:3) experienced a resuscitated out of hospital cardiac arrest. MRI revealed normal left and right ventricular indexed dimensions and function, with no evidence of myocardial fibrosis, and no features of cardiomyopathy. She was found to have idiopathic ventricular fibrillation and an ICD was implanted, which has subsequently delivered two appropriate shocks (Figure 3). She has responded successfully to quinidine therapy. Her sister (III:2), age 29 years, has been repeatedly evaluated and all cardiovascular investigations have been normal, including a normal ECG, echocardiogram, electrophysiological study and a 7-day Holter monitor. Clinical data, including cardiac screening results, were not available for the proband’s grandparents (I:I and I:2) and uncle (II:1).

Ventricular fibrillation in relative III:3. Example of a short coupled ventricular ectopic triggering VF, as is typically seen in idiopathic ventricular fibrillation.
Exome sequencing
To identify a pathogenic mutation responsible for the cardiac phenotype in family ALB, exome sequencing was performed on the proband’s DNA. 62,859,256 sequence reads of 101 bp were generated, of which 38,964,387 mapped uniquely to target regions, representing an average exome-wide coverage of 50.7 reads, with 82.2% of target bases covered at least 20 times. 44,508 SNVs and 4,837 InDels were identified across the 62 Mb target exome, of which 8,214 SNVs caused a non-synonymous, splice-site or nonsense change and 410 InDels occurred in the coding regions or splice signal sequences. We excluded all homozygous variants and heterozygous variants present at >1% frequency in any ethnic subgroup of the 1000 Genomes Project data, or in 6500 exomes of ESP, leaving 259 rare non-synonymous, splice site and nonsense SNVs in 250 genes (Additional file 2) and 13 rare in-frame, frameshift and splice site InDels in 13 genes (Additional file 3). Genes were sorted by expression level in cardiac tissue, derived from RNAseq data, which ranked ACTN2 Ala119Thr non-synonymous variation as number 1; the top 20 ranked variations are shown in Table 2.
Genotyping and haplotype analysis
Three non-synonymous variants in cardiac disease-associated genes were selected for co-segregation analysis in family members as they affected conserved residues in genes associated with primary cardiac pathologies: Ala119Thr in ACTN2, previously reported in a family with heterogeneous HCM [14], Ala572Asp in SCN5A (rs36210423), and Asp26Asn in plakophillin 2 (PKP2) (rs143004808). Sanger DNA sequencing confirmed the presence of each variant in the proband’s DNA, however, only Ala119Thr in ACTN2 co-segregated with disease in all of the affected family members, and was also present in one clinically unaffected female (III:2; Figure 4A, B).

Genotyping and haplotype analysis in ALB and EI families. (A) Haplotype analysis in the current Family ALB, (B) DNA sequences of the haplotype, and (C) haplotype analysis in previous Family EI. *inferred haplotype.
Since we had previously identified the ACTN2 Ala119Thr variant in an apparently unrelated Australian family with clinically heterogeneous HCM [14], we performed haplotype analysis to determine if the variant segregates on the same haplotype in both families. Five VNTRs spanning a 2.9 Mb region around ACTN2 were selected; D1S2850 and D1S2670, as used in our previous study, plus one VNTR each in ACTN2 and RYR2, and D1S2678. Haplotype analysis revealed that the Ala119Thr variant segregates with a unique haplotype common to both families, defined by allele sizes of 259 bp in D1S2678, 155 bp in D1S2850, 142 bp in the ACTN2 VNTR, 193 bp in the RYR2 VNTR and 118 bp in D1S2670 (Figure 4A, C). We previously determined that the Ala119Thr variant was absent in 297 unrelated probands with HCM, however, since LVNC was a feature in both ALB and EI families, we genotyped a panel of 31 unrelated patients with LVNC. The Ala119Thr variant was absent in all samples (not shown).
Discussion
Clinical evaluation of an Australian family revealed diverse cardiac pathologies in four affected members and genetic testing of the exome identified a pathogenic ACTN2 heterozygous variant (Ala119Thr) that co-segregated with disease. The variant affects an amino acid in the highly conserved actin-binding CH1 domain [14]. The diverse clinical phenotypes seen in this family, including idiopathic ventricular fibrillation, LVNC, and sudden unexplained death, suggest mutations in the ACTN2 gene likely perturb a number of different mechanical and arrhythmogenic substrates. In families with such variable clinical presentations, a non-biased whole exome sequencing approach may be useful in elucidating the underlying genetic cause of disease.
We previously identified the same novel mutation as the cause of a marked clinically heterogeneous cardiac phenotype in a 3 generation family with 27 members (EI family) using genome-wide linkage analysis [14]. The Ala119Thr mutation is absent from 260 control samples, 6500 ESP exomes and the 1000 Genomes data, and segregates with the same unique haplotype in the ALB and EI families. The variant segregates with all affected members of both families (n = 11). Collectively this further validates the pathogenicity of the Ala119Thr mutation in ACTN2, which is likely inherited from a common ancestor of both families.
The diversity of clinical phenotypes is a major fascination both in the current (ALB) and our previously reported (EI) families. The spectrum of clinical presentations and clinical outcomes is remarkably similar in both families, and includes both structural and arrhythmogenic pathologies. Several individuals have either hypertrophic cardiomyopathy (both classical and apical forms) or isolated LVNC. Cardiac arrhythmogenic abnormalities, including atrial and ventricular fibrillation, resuscitated cardiac arrest, and appropriate ICD discharges have been reported in a number of individuals in both families. Furthermore, there have been two sudden cardiac deaths in young people aged under 35 years, one where hypertrophic cardiomyopathy was known, and one in which no cause was identified at post-mortem. Heart transplantation for end-stage heart failure has occurred in one individual, presumably due to “burnt-out” hypertrophic cardiomyopathy.
While phenotypic heterogeneity and incomplete penetrance are common features of inherited cardiac diseases, it is unclear how divergent cardiac phenotypes emerge from the same mutation, and so exemplified by the structural and arrhythmogenic pathologies caused by the Ala119Thr mutation in ACTN2. The ACTN2 protein primarily functions to anchor and crosslink actin filaments in the cardiac Z-disc at the lateral boundaries of the sarcomere [15]. The Z-disc provides structural support, by tethering the sarcomere to the sarcolemma via the costameres, and by anchoring filamentous F-actin, titin and nebulette [16]. Depletion of actn2 expression in zebrafish embryos, using antisense oligomers, results in a disruption of the lateral alignment of discs [17]. In humans, analysis of a missense and nonsense mutation in the ACTN2 interacting protein myopalladin [18], revealed a mutation dependent disruption to myofibrillogenesis, or abnormal assembly of terminal Z-discs [19]. Therefore, ACTN2 and the Z-disc can be regarded as a scaffold. Additionally, the Z-disc serves as an interaction platform for proteins that shuttle to the nucleus, such as calcineurin, ankyrin repeat domain 2 and cardiac ankyrin repeat protein, and they may represent molecular messengers that translate mechanical stress into a transcriptional response [20]-[23]. Therefore the Z-disc can also be regarded as a signalling network that is well positioned to sense mechanical stress [20],[24].
Furthermore, there is evidence of ACTN2 directly interacting with cardiac ion channels, such as the potassium ion channels KCNA4 and KCNA5[25],[26], the sodium ion channel SCN5A[27], and it forms a bridge between the calcium ion channels CACNA1C and CACNA1D[28]. Thus, disruption of ACTN2 may impact on the localisation and function of cardiac ion-channels. It is tempting to speculate that the different clinical presentations of Ala119Thr result from a stochastic disruption to one of the many functional roles of ACTN2 (Figure 5).

Alpha-actinin2 and cardiac disease. Potential mechanisms by which mutations in the ACTN2 gene can lead to diverse cardiac phenotypes.
The precedent of mutations in a single gene leading to diverse clinical phenotypes has been documented in other settings. Most notably, mutations in the lamin A/C (LMNA) gene have been identified in a diverse range of clinical phenotypes. Originally described in Emery Dreifuss muscular dystrophy, mutations in the lamin A/C gene have now been identified in a number of other diverse diseases including familial dilated cardiomyopathies with conduction disease, limb girdle muscular dystrophies, lipodystrophies, Hutchinson-Gilford progeria syndrome, and mandibuloacral dysplasia, collectively called the “laminopathies” [29],[30]. Like the multifaceted role of lamins, and their impact on various biological signalling pathways and functions, the ACTN2 protein has similar diversity in its potential mechanistic roles (Figure 5), and may contribute to a collection of these diseases as “Z-discopathies”.
The current study highlights the emerging role of exome sequencing in the identification of a causative gene mutation. The traditional approach of using the clinical phenotype to select the appropriative genetic testing panel would have required consideration of a cardiomyopathy, sudden death, or arrhythmogenic gene panel, based on the diverse phenotype of the family. ACTN2 is unlikely to be included on an arrhythmogenic gene diagnostic panel or a common cardiomyopathy gene panel in which each gene is responsible for >5% of disease. However, ACTN2 may be included on comprehensive cardiomyopathy gene panel that includes minor disease-associated genes. Exome sequencing to investigate the genetic basis of disease in this family was non-biased, robust, and comprehensive, and provided the broadest coverage of possible disease genes to facilitate the identification of the causative mutation. A number of studies have recently emerged demonstrating the use of exome sequencing in genetic discoveries in both primary arrhythmogenic disorders and inherited cardiomyopathies, and therefore provides an exciting new approach to disease gene discovery in cardiovascular disease [31]-[34].
Conclusions
Cardiac phenotype heterogeneity is a clinical challenge within families with inherited heart disease. Understanding the underlying genetic causes has major implications both in the diagnosis and screening of at-risk family members, and in shedding light on the mechanisms that underpin disease pathogenesis. The mutation identified in the ACTN2 gene is likely ancestral to both ALB and EI families and is responsible for the marked cardiac phenotype heterogeneity observed in family members, including both structural and arrhythmogenic abnormalities, including sudden death. The diverse mechanistic roles of ACTN2 in the cardiac Z-disc may explain this heterogeneous clinical presentation. The newer approach of exome sequencing is a useful adjunct to cardiac genetic testing in families with mixed clinical presentations.
Additional files
Abbreviations
- BrS:
-
Brugada syndrome
- DCM:
-
Dilated cardiomyopathy
- HCM:
-
Hypertrophic cardiomyopathy
- LQTS:
-
Long QT syndrome
- LVNC:
-
Left ventricular non-compaction
- SQTS:
-
Short QT syndrome
- IVF:
-
Idiopathic ventricular fibrillation
- SNV:
-
Single nucleotide variation
- ICD:
-
Implantable cardioverter defibrillator
References
Wilde AA, Behr ER: Genetic testing for inherited cardiac disease. Nat Rev Cardiol. 2013, 10 (10): 571-583. 10.1038/nrcardio.2013.108.
Maron BJ, Maron MS, Semsarian C: Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol. 2012, 60 (8): 705-715. 10.1016/j.jacc.2012.02.068.
Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG: A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell. 1990, 62 (5): 999-1006. 10.1016/0092-8674(90)90274-I.
Karam S, Raboisson MJ, Ducreux C, Chalabreysse L, Millat G, Bozio A, Bouvagnet P: A de novo mutation of the beta cardiac myosin heavy chain gene in an infantile restrictive cardiomyopathy. Congenit Heart Dis. 2008, 3 (2): 138-143. 10.1111/j.1747-0803.2008.00165.x.
Villard E, Duboscq-Bidot L, Charron P, Benaiche A, Conraads V, Sylvius N, Komajda M: Mutation screening in dilated cardiomyopathy: prominent role of the beta myosin heavy chain gene. Eur Heart J. 2005, 26 (8): 794-803. 10.1093/eurheartj/ehi193.
Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, Ortiz-Lopez R, Wang Z, Antzelevitch C, O'Brien RE, Schulze-Bahr E, Keating MT, Towbin JA, Wang Q: Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998, 392 (6673): 293-296. 10.1038/32675.
Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT: SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995, 80 (5): 805-811. 10.1016/0092-8674(95)90359-3.
Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, Camm AJ, Ellinor PT, Gollob M, Hamilton R, Hershberger RE, Judge DP, Le Marec H, McKenna WJ, Schulze-Bahr E, Semsarian C, Towbin JA, Watkins H, Wilde A, Wolpert C, Zipes DP: HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace. 2011, 13 (8): 1077-1109. 10.1093/europace/eur245.
Meder B, Haas J, Keller A, Heid C, Just S, Borries A, Boisguerin V, Scharfenberger-Schmeer M, Stahler P, Beier M, Weichenhan D, Strom TM, Pfeufer A, Korn B, Katus HA, Rottbauer W: Targeted next-generation sequencing for the molecular genetic diagnostics of cardiomyopathies. Circ Cardiovasc Genet. 2011, 4 (2): 110-122. 10.1161/CIRCGENETICS.110.958322.
Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, Hardison M, Person R, Bekheirnia MR, Leduc MS, Kirby A, Pham P, Scull J, Wang M, Ding Y, Plon SE, Lupski JR, Beaudet AL, Gibbs RA, Eng CM: Clinical Whole-Exome Sequencing for the Diagnosis of Mendelian Disorders. N Engl J Med. 2013, 369 (16): 1502-11. 10.1056/NEJMoa1306555.
Li H, Durbin R: Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010, 26 (5): 589-595. 10.1093/bioinformatics/btp698.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R: The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009, 25 (16): 2078-2079. 10.1093/bioinformatics/btp352.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA: The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20 (9): 1297-1303. 10.1101/gr.107524.110.
Chiu C, Bagnall RD, Ingles J, Yeates L, Kennerson M, Donald JA, Jormakka M, Lind JM, Semsarian C: Mutations in alpha-actinin-2 cause hypertrophic cardiomyopathy: a genome-wide analysis. J Am Coll Cardiol. 2010, 55 (11): 1127-1135. 10.1016/j.jacc.2009.11.016.
Sjoblom B, Salmazo A, Djinovic-Carugo K: Alpha-actinin structure and regulation. Cell Mol Life Sci. 2008, 65 (17): 2688-2701. 10.1007/s00018-008-8080-8.
Luther PK: The vertebrate muscle Z-disc: sarcomere anchor for structure and signalling. J Muscle Res Cell Motil. 2009, 30 (5-6): 171-185. 10.1007/s10974-009-9189-6.
Yang J, Xu X: alpha-Actinin2 is required for the lateral alignment of Z discs and ventricular chamber enlargement during zebrafish cardiogenesis. FASEB J. 2012, 26 (10): 4230-4242. 10.1096/fj.12-207969.
Bang ML, Mudry RE, McElhinny AS, Trombitas K, Geach AJ, Yamasaki R, Sorimachi H, Granzier H, Gregorio CC, Labeit S: Myopalladin, a novel 145-kilodalton sarcomeric protein with multiple roles in Z-disc and I-band protein assemblies. J Cell Biol. 2001, 153 (2): 413-427. 10.1083/jcb.153.2.413.
Purevjav E, Arimura T, Augustin S, Huby AC, Takagi K, Nunoda S, Kearney DL, Taylor MD, Terasaki F, Bos JM, Ommen SR, Shibata H, Takahashi M, Itoh-Satoh M, McKenna WJ, Murphy RT, Labeit S, Yamanaka Y, Machida N, Park JE, Alexander PM, Weintraub RG, Kitaura Y, Ackerman MJ, Kimura A, Towbin JA: Molecular basis for clinical heterogeneity in inherited cardiomyopathies due to myopalladin mutations. Hum Mol Genet. 2012, 21 (9): 2039-2053. 10.1093/hmg/dds022.
Frank D, Kuhn C, Katus HA, Frey N: The sarcomeric Z-disc: a nodal point in signalling and disease. J Mol Med (Berl). 2006, 84 (6): 446-468. 10.1007/s00109-005-0033-1.
Frey N, Richardson JA, Olson EN: Calsarcins, a novel family of sarcomeric calcineurin-binding proteins. Proc Natl Acad Sci U S A. 2000, 97 (26): 14632-14637. 10.1073/pnas.260501097.
Kojic S, Medeot E, Guccione E, Krmac H, Zara I, Martinelli V, Valle G, Faulkner G: The Ankrd2 protein, a link between the sarcomere and the nucleus in skeletal muscle. J Mol Biol. 2004, 339 (2): 313-325. 10.1016/j.jmb.2004.03.071.
Zou Y, Evans S, Chen J, Kuo HC, Harvey RP, Chien KR: CARP, a cardiac ankyrin repeat protein, is downstream in the Nkx2-5 homeobox gene pathway. Development. 1997, 124 (4): 793-804.
Frank D, Frey N: Cardiac Z-disc signaling network. J Biol Chem. 2011, 286 (12): 9897-9904. 10.1074/jbc.R110.174268.
Cukovic D, Lu GW, Wible B, Steele DF, Fedida D: A discrete amino terminal domain of Kv1.5 and Kv1.4 potassium channels interacts with the spectrin repeats of alpha-actinin-2. FEBS Lett. 2001, 498 (1): 87-92. 10.1016/S0014-5793(01)02505-4.
Maruoka ND, Steele DF, Au BP, Dan P, Zhang X, Moore ED, Fedida D: alpha-actinin-2 couples to cardiac Kv1.5 channels, regulating current density and channel localization in HEK cells. FEBS Lett. 2000, 473 (2): 188-194. 10.1016/S0014-5793(00)01521-0.
Ziane R, Huang H, Moghadaszadeh B, Beggs AH, Levesque G, Chahine M: Cell membrane expression of cardiac sodium channel Na(v)1.5 is modulated by alpha-actinin-2 interaction. Biochemistry. 2010, 49 (1): 166-178. 10.1021/bi901086v.
Lu L, Zhang Q, Timofeyev V, Zhang Z, Young JN, Shin HS, Knowlton AA, Chiamvimonvat N: Molecular coupling of a Ca2 + −activated K + channel to L-type Ca2+ channels via alpha-actinin2. Circ Res. 2007, 100 (1): 112-120. 10.1161/01.RES.0000253095.44186.72.
Worman HJ: Nuclear lamins and laminopathies. J Pathol. 2012, 226 (2): 316-325. 10.1002/path.2999.
Schreiber KH, Kennedy BK: When lamins go bad: nuclear structure and disease. Cell. 2013, 152 (6): 1365-1375. 10.1016/j.cell.2013.02.015.
Marsman RF, Barc J, Beekman L, Alders M, Dooijes D, Van-den-Wijngaard A, Ratbi I, Sefiani A, Bhuiyan ZA, Wilde AA, Bezzina CR: A mutation in CALM1 encoding calmodulin in familial idiopathic ventricular fibrillation in childhood and adolescence. J Am Coll Cardiol. 2013, 63 (3): 259-66. 10.1016/j.jacc.2013.07.091.
Crotti L, Johnson CN, Graf E, De Ferrari GM, Cuneo BF, Ovadia M, Papagiannis J, Feldkamp MD, Rathi SG, Kunic JD, Pedrazzini M, Wieland T, Lichtner P, Beckmann BM, Clark T, Shaffer C, Benson DW, Kääb S, Meitinger T, Strom TM, Chazin WJ, Schwartz PJ, George AL: Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation. 2013, 127 (9): 1009-1017. 10.1161/CIRCULATIONAHA.112.001216.
Norton N, Li D, Rampersaud E, Morales A, Martin ER, Zuchner S, Guo S, Gonzalez M, Hedges DJ, Robertson PD, Krumm N, Nickerson DA, Hershberger RE: Exome sequencing and genome-wide linkage analysis in 17 families illustrate the complex contribution of TTN truncating variants to dilated cardiomyopathy. Circ Cardiovasc Genet. 2013, 6 (2): 144-153. 10.1161/CIRCGENETICS.111.000062.
Norton N, Li D, Rieder MJ, Siegfried JD, Rampersaud E, Zuchner S, Mangos S, Gonzalez-Quintana J, Wang L, McGee S, Reiser J, Martin E, Nickerson DA, Hershberger RE: Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am J Hum Genet. 2011, 88 (3): 273-282. 10.1016/j.ajhg.2011.01.016.
Acknowledgements
CS is the recipient of a National Health and Medical Research Council (NHMRC) Practitioner Fellowship (#571084). This study was also supported, in part, by an NHMRC project grant (#632575).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RDB contributed to study design, acquisition of data, analysis and interpretation of data and drafting the manuscript. LKM and JMK contributed to acquisition of data and drafting the manuscript. CS contributed to study design, acquisition of data, analysis and interpretation of data and drafting the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12881_2014_99_MOESM1_ESM.xlsx
Additional file 1: Table S1.: Primer sequences, and PCR amplification conditions for microsatellite markers. (XLSX 10 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Bagnall, R.D., Molloy, L.K., Kalman, J.M. et al. Exome sequencing identifies a mutation in the ACTN2 gene in a family with idiopathic ventricular fibrillation, left ventricular noncompaction, and sudden death. BMC Med Genet 15, 99 (2014). https://doi.org/10.1186/s12881-014-0099-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-014-0099-0