
Abstract
Background
Soil phosphorus (P) deficiency is one of the major limiting factors to crop production. The development of crop varieties with improved P use efficiency (PUE) is an important strategy for sustainable agriculture. The objectives of this research were to identify quantitative trait loci (QTLs) linked to PUE traits using a high-density single nucleotide polymorphism (SNP) map and to estimate the epistatic interactions and environmental effects in rice (Oryza sativa L.).
Results
We conducted a two-year field experiment under low and normal P conditions using a recombinant inbred population of rice derived from Zhenshan 97 and Minghui 63 (indica). We investigated three yield traits, biomass (BIOM), harvest index (HI), and grain yield (Yield), and eight PUE traits: total P uptake (PUP), P harvest index (PHI), grain P use efficiency (gPUE) based on P accumulation in grains, straw P use efficiency (strPUE) based on P accumulation in straw, P use efficiency for biomass (PUEb) and for grain yield (PUEg) based on P accumulation in the whole plant, P translocation (PT), and P translocation efficiency (PTE). Of the 36 QTLs and 24 epistatic interactions identified, 26 QTLs and 12 interactions were detected for PUE traits. The environment affected seven QTLs and three epistatic interactions. Four QTLs (qPHI1 and qPHI2 for PHI, qPUEg2 for PUEg, and qPTE8 for PTE) with strong effects were environmentally independent. By comparing our results with similar QTLs in previous studies, three QTLs for PUE traits (qPUP1 and qPUP10 for PUP, and qPHI6 for PHI) were found across various genetic backgrounds. Seven regions were shared by QTLs for yield and PUE traits.
Conclusion
Most QTLs linked to PUE traits were different from those linked to yield traits, suggesting different genetic controls underlying these two traits. Those chromosomal regions with large effects that are not affected by different environments are promising for improving P use efficiency. The seven regions shared by QTLs linked to yield and PUE traits imply the possibility of the simultaneous improvement of yield and PUE traits.
Similar content being viewed by others

Background
Phosphorus (P) is an important nutrient for crops. Low levels of available P in soils and P use efficiency (PUE) are becoming the two major constraints for crop production. The addition of P to soils increases crop production costs, exhausts non-renewable P resources, and causes environmental problems [1]-[3]. Therefore, it is desirable to develop cultivars with high PUE.
Phosphorus use efficiency is improved by increasing both P uptake and use efficiencies [3],[4]. For crops, P deficiency may be attributed to low total P content or low available P in soils; with respect to the latter, soils contain considerable amounts of P, but a large proportion is soil bound or in the organic form, and thus cannot be utilized by plants [5]. Therefore, improving PUE can be approached by various strategies. For soils with low total P content, strategies include regular applications of small doses of P and the improvement of internal PUE. Under P deficiency, crops have the ability to obtain more P through increasing the activities of enzymes involved in P scavenging and recycling and by altering respiratory electron transport and other metabolic pathways [1],[6]. For soils with high unavailable P content, the most common strategy is the increase in P uptake efficiency through the proliferation and extension of plant roots, including the selection of deep rooting, thick roots, and strong root penetration ability [5]. Most investigations have focused on increasing P acquisition abilities by improving features such as root traits, root exudates, mycorrhiza, and high-affinity P transporters [7]-[10].
Among several parameters often investigated for P acquisition ability, P uptake (PUP) is an integrative trait that directly reflects a plant’s ability to acquire P. For example, Wissuwa et al. [3] used PUP to estimate P uptake efficiency and identified a major quantitative trait locus (QTL) named Pup1 on chromosome 12 of rice. Further studies have shown that Nipponbare near-isogenic lines carrying Pup1 could increase PUP in a severely P-deficient field, relative to the recurrent parent Nipponbare [11]. The high yield of modern rice varieties is mainly attributed to the high harvest index and great PUE for grain yield, which are due to the plants’ ability to mobilize P from vegetative to reproductive tissues [12]. However, the genetic relationship between grain yield formation and the traits associated with P uptake, re-translocation, and partitioning in plants have rarely been reported in previous studies [13],[14].
Genome mapping can be used to locate the QTLs linked to PUE traits, which are controlled by multiple genes and show the genetic characteristics of quantitative traits [8],[15],[16]. In recent decades, QTLs for PUE and tolerance to P deficiency have been identified in rice [3],[17], wheat [18], maize [19],[20], and soybean [21],[22]. Those studies mainly used two approaches. First, they focused on indirect traits, such as relative growth, relative tiller, root traits, shoot dry weight, and relative yield. Second, they investigated trait performance under P deficient conditions [8],[17],[23],[24]. However, traits directly related to PUE, such as PUE for grain yield (PUEg), P harvest index (PHI), and P translocation efficiency (PTE), which are based on P uptake, grain yield, and biomass production, have rarely been used for evaluating and mapping QTLs [4],[18],[19].
Although dozens of QTLs have been identified in a single growing season in plants experiencing P-starvation, few QTLs have been detected in different years at the same or different locations, or in different genetic populations. Hence, as most traits are profoundly influenced by many genes and show various genotypes due to environmental interactions, few QTLs can be used to improve PUE [25]. Those QTLs detected for PUE traits or tolerance to P deficiency may be specifically functional or only fully expressed in a given environment. Furthermore, most previous investigations were carried out under controlled conditions or based on small crop populations with small experimental field plots [3],[18],[26]. Bray [27] emphasized that controlled conditions can never fully mimic field scenarios because crop plants grown in the field may face multiple abiotic and biotic factors. Therefore, more investigations on QTLs linked to PUE traits should be performed in several different environments.
Most QTL identifications in previous studies were based on linkage maps using restriction fragment length polymorphism (RFLP) and simple sequence repeat (SSR) markers. In those maps, sparse markers in many regions made it impossible to obtain precise and complete information about the number and locations of the QTLs [28]. The QTLs for PUE traits and P-deficiency tolerance in the previous studies were often located over a large confidence interval in RFLP/SSR maps, which complicated identification when there were several minor QTLs closely linked in the interval. Recently, new markers, such as single feature polymorphism (SFP) [29] and single nucleotide polymorphism (SNP) [28],[30],[31], have been used for QTL identification. These methods have facilitated the construction of high-density genetic maps, and have allowed precise and effective detections of QTLs in soybean, rice, and maize.
In most pervious reports, PUP was often used for estimating the P acquisition efficiency from soils; however PUEg or PUE for total biomass (PUEb) at maturity are better indicators in terms of biomass or grain yield. To investigate the physiological mechanisms of PUE, Rose and Wissuwa [32] suggested that it is preferable to dissect PUE into plant components, such as grain PUE (gPUE) and shoot PUE (strPUE). In addition, there is also significant P remobilization from leaves and stems during grain development to the developing grains [13]. Moreover, PHI is considered a parameter for PUE [33]. These three parameters, i.e., PHI, P translocation from stems to grains (PT), and P translocation efficiency (PE), are used to reflect P translocation from stems to grains during grain filling, which is associated with grain yield. In this study, a recombinant inbred population derived from a cross between Zhenshan 97 and Minghui 63, along with a high-density SNP bin map from Yu et al. [28], were used to locate QTLs linked to yield and eight PUE traits under two P application rates. The main objectives of the present study were (1) to locate QTLs for PUE traits and (2) to investigate the genetic relationship between PUE traits and yield traits and their stability across environments.
Methods
Plant materials and field experiments
The recombinant inbred line (RIL) population used in the study was derived by single-seed descent from a cross between two elite rice lines of indica subspecies, Zhenshan 97 and Minghui 63, the parents of Shanyou 63, the most widely cultivated hybrid in China [28],[34]. Lines with too short a growth duration and with very low grain yield were not used for the field experiments because it is less significant to estimate PUE traits based on low grain yield or biomass from the viewpoint of crop production. Additionally, our experiment investigated PUE traits in large plots under real conditions for rice production. Thus, a total of 113 lines, plus the two parents, were used for the field experiments in 2008 and 2009.
The field experiments were carried out in the farmers’ field in Dajin town, Wuxue city, Hubei province, China (29°51' N, 115°33' E) during the rice-growing season from May to October in 2008 and 2009. The soil type was gleyed paddy soil, and it exhibited the following properties in the top 25 cm: pH 5.20, 25.89 g kg−1 organic C, 1.57 g kg−1 total N, 5.35 mg kg−1 available Olsen-P, and 54.93 mg kg−1 exchangeable K.
The experiments were conducted following a randomized complete block design with three replicates. Each replicate contained two P application rates: low P (without P fertilizer) and normal P applications (with pure P of 40 kg ha−1, equal to 92 kg P2O5 ha−1). All the P was applied as basal fertilizer in the form of calcium superphosphate one day before transplanting. To support high grain yield, a total of 135 kg N ha−1 in the form of urea was applied three times: 54 (40%) kg ha−1 as basal fertilizer, 40.5 (30%) kg ha−1 15 days after transplanting (DAT), and 40.5 (30%) kg ha−1 25 DAT. Potassium (100 kg K ha−1 as potassium chloride) was applied two times: 50 kg ha−1 as basal fertilizer and 50 kg ha−1 25 DAT. Zinc (5 kg Zn ha−1) was applied in the form of zinc sulfate heptahydrate as basal fertilizer. Under the low P application, the applications of the other fertilizers were the same as those under the normal P application. All the fertilizers were applied during an early growth stage.
In both years, seeds were sown in nursery plastic plates on 17 May and the seedlings were transplanted on 15 June. Each line was transplanted to plots with a spacing of 0.20 × 0.17 m and an area of 8.2 m2. Each plot included 14 rows with 16 hills per row and three 27-day-old seedlings per hill. To minimize seepage between the P applications, the main plots were separated with double bunds to prevent water flow, and all the bunds were covered with plastic film, extending to a depth of 20 cm below the soil surface. To avoid loss and movement of the fertilizers, the plots were not drained during the duration of the experiment. A flood-irrigation system was adopted, which followed high-yield agricultural practices according to the local rice production. Pests, diseases, birds, and weeds were intensively controlled.
Sampling and measurements
At the heading of the plants from each line (plot), eight uniform plants were sampled (excluding the border plants). The plants were separated into leaves and stems (including culms, sheaths, and young panicles). At maturity, twelve uniform plants were harvested from the middle of each plot. All the panicles were collected and hand-threshed, and then all the grains were divided into filled and unfilled groups by submerging them in tap water. All the leaves, stems (culms and sheaths), rachis, and filled and unfilled grains were separately collected and oven-dried at 80°C until a constant weight was achieved. The grain yield (Yield, g m−2) was reported at a 14% moisture content basis. The harvest index (HI, %) was calculated as the ratio of grain dry matter to total aboveground biomass (BIOM, g m−2). The BIOM, Yield, and HI were considered as yield traits in the study.
Measurements of P concentration
The oven-dried leaves, stems, and filled grains were separately grinded into powders and mixed thoroughly, and each powder was passed through a 1-mm sieve. Approximately 0.2 g of each sample powder was digested with sulfuric acid and hydrogen peroxide to determine the P concentration spectrophotometrically according to the molybdenum blue method [35], using a continuous-flow analyzer (FUTURA, Alliance Instrument, France). The P concentration was calculated based on dry weight.
Definitions of PUE traits
Eight PUE traits were calculated, as described by Dordas [13], Jones et al. [33], and Rose and Wissuwa [32]. The P accumulation of each plant part (including leaves, stems, and grains) is the product of the part’s dry weight and its corresponding P concentration. The P uptake at maturity (PUP, g m−2) is the sum of the accumulations in the various plant parts per m2. The P harvest index (PHI, %) is the ratio of the grain P accumulation to the total P accumulation of the aboveground parts at maturity. The grain PUE (gPUE, g g−1) is defined as the filled grain dry weight per g P in grains. The straw PUE (strPUE, g g−1) is defined as the straw dry weight per g P in straw at maturity. The PUE for biomass (PUEb, g g−1) is defined as the total aboveground biomass per g P accumulated in the whole plant at maturity. The PUE for grain yield (PUEg, g g−1) is defined as the grain yield per g P accumulated in the whole plant at maturity. The P translocation from stems to grains (PT, g m−2) is calculated as the leaf and stem P accumulations at heading minus those at maturity. The P translocation efficiency (PTE, %) is defined as the ratio of PT to P accumulation at heading. A network diagram for the investigated PUE traits are presented in Figure 1.

The three yield traits and eight P use efficiency traits and their inter-relationships. gPUE: P use efficiency for grain yield based on P accumulation in grains, PT: P translocation, PTE: P translocation efficiency, PUP: total aboveground P uptake, PUEb: P use efficiency for biomass accumulation, PUEg: P use efficiency for grain yield, strPUE: P use efficiency for straw dry weight based on P accumulation in straws. represents a positive relationship between two traits, represents a negative relationship, represents no obvious relationship.
Phenotype data analysis
The means over three replicates were used for all the statistical analyses, which were conducted using SAS 9.1 (North Carolina, USA). The broad-sense heritability (h B 2, %) based on the RILs was estimated according to the following formula: h B 2 = σ 2 g /(σ 2 g + σ 2 ge /e + σ 2 e /re), where r is the number of replicates per year, e is number of environments (years), σ 2 g is the genetic variance, σ 2 ge is the variance of genotype due to environmental interactions, and σ 2 e is the residual variance [36].
Construction of genetic linkage map and QTL detection
A high-density bin map based on SNP, as described by Xie et al. [37] and Yu et al. [28], was constructed. The map consisted of 1619 recombination bins, covering all chromosomes without missing data and spanning 1625.5 cM in length, with an average interval of 1.0 cM between adjacent SNP markers. All the markers were used for the QTL mapping analysis in this study. Due to the large number of SNP markers, the markers not involved in candidate QTLs or epistatic interactions were removed from the genetic map figures.
As revealed by the analysis of variance, the different P applications (low and normal) and study years (2008 and 2009) had different effects on the traits. Thus, the four combinations of these factors (two years and two P applications) were considered as four environmental factors (e1 and e2 represented low P in 2008 and 2009; e3 and e4 represented normal P in 2008 and 2009). The mean of three replicates of each P application and each year were considered as the phenotypic score for each environment. All of the phenotype data were normally distributed and directly used for the QTL detection without any transformations. The QTL detection was performed with the QTLNetwork-2.0 software (Institute of bioinformatics, Zhejiang University, Hangzhou, China, Yang and Zhu 2005) based on a mixed linear model [38]. Composite interval analyses were conducted with a 10 cM window size and a 0.5 cM walking speed. One thousand permutations were performed for each trait to calculate a critical F value at P < 0.05. The Monte Carlo Markov Chain was applied to estimate the QTL effects. A QTL was declared if the phenotype was associated with a marker locus at P < 0.005. The QTL naming followed the procedure presented by McCouch et al. [39]. Additive × additive effects (aa), additive × environment interactions (ae), and additive × additive × environment interactions (aae) were separately estimated using the QTL location software.
Individual and multi-environment (e1, e2, e3, and e4) combined analyses were performed, and the locations and effects of the QTLs were compared between individual and combined analyses. The locations and effects of QTLs were reported, as well as the epistatic interactions detected by the combined analysis across the four environments. Additionally, we also clarified the individual environments for which similar QTLs for a given trait were identified in the same or neighboring region by individual environment analysis.
Results
Phenotypic variation
Under both low and normal P conditions in both two years, Minghui 63 had higher BIOM, Yield, PUP, PUEb, and PT, but lower HI, PHI, PUEg, and PTE than Zhenshan 97 (Table 1). With the exception of PUEg and PT for Zhenshan 97 under low P, the two parents had higher BIOM, HI, Yield, PUP, PHI, PUEg, PT, and PTE in 2008 than in 2009 under both P applications. Under low P, the PHI, gPUE, strPUE, PUEb, PUEg, and PTE of the two parents were higher than under normal P in both years. Minghui 63 had a higher PT under low P compared with under normal P; however, the opposite was true for Zhenshan 97.
All the traits varied widely under both the low and normal P applications in both years (Table 1), and the transgressive variations were both positive and negative. The broad-sense heritabilities for all 11 traits varied widely, ranging from 54.9% for gPUE to 90.9% for BIOM under low P, and from 58.7% for PT to 86.9% for BIOM under normal P (Table 1). Generally, BIOM, PUP, and PTE had high heritabilities, whereas gPUE, PUEg, and PT showed low heritabilities. The 11 traits varied significantly with year, P level, and genotype (Table 2).
Correlation among various traits
For all traits, significant positive correlations were observed between low and normal P applications in 2008. Similar correlations were also found in 2009. These correlations ranged from 0.40 for gPUE to 0.90 for both Yield and HI in 2008, and from 0.64 for PUEb to 0.93 for HI in 2009 (Table 3). Significant positive correlations were also found between all trait values from 2008 to 2009 under both P applications.
The main inter-relationships among the 11 investigated traits are presented Figure 1. Generally, three yield traits were significantly correlated with PUE traits (Table 3). There were similar correlations among the eight PUE traits under low and normal P applications in both years, separately. Under both the low and normal P applications, PUP was negatively correlated with strPUE, PUEb, and PTE in each year; however, PUP was negatively correlated with PHI and PUEg in 2009 only. Moreover, PHI was positively correlated with strPUE, PUEb, PUEg, PT, and PTE. Under the two P applications, gPUE was positively correlated with PUEb and PUEg in each year. Positive correlations between strPUE and PUEb and between PUEg and PTE under the two P applications in each year were found. Under the two P applications in each year, PUEb was positively correlated with PUEg and PTE, and PUEg was positively correlated with PT and PTE.
QTL detection
Based on the multi-environment combined analysis, a total of 36 QTLs were detected for the 11 investigated traits (Table 4), 26 of which also detected in the similar regions by individual environment analysis. Compared with the QTL locations determined by individual environment analysis, seven of the 36 QTLs were simultaneously detected in both years, 14 were simultaneously detected in different P applications, and 16 were located in 2 or more individual environments.
QTLs for BIOM
Five QTLs for BIOM were detected, and they explained 34.0% of the total phenotypic variation (Table 4 and Figure 2). Minghui 63, the parent with a high BIOM value, contributed alleles at three QTLs (qBIOM7, qBIOM10, and qBIOM11), and Zhenshan 97 provided two alleles at the other two QTLs. Two QTLs (qBIOM2 and qBIOM7) exhibited interactions with the e4 environmental factor (normal P in 2009), and each interaction explained 0.5% and 0.4% of the total variation, respectively. Except for qBIOM2, the remaining four QTLs were detected in two environments across two years.

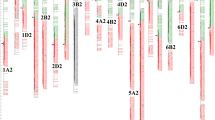
A genetic linkage map of rice showing the mapping of QTLs with additive effects and epistatic effects. The sequent SNP markers have been sparsed according to the mapping results. The filled symbols represent the QTLs with additive effects; the open symbols represent the non-QTL locations involved in epistatic interactions. indicate the QTLs or location detected for BIOM; for HI; for Yield; for PUP; for PHI; for gPUE; for strPUE; for PUEb; for PUEg; for PT; and for PTE. Markers with arrows indicate a QTL located in a similar region according to RFLP/SSR maps and physical positions in previous studies. Marker RM259 on chromosome 1 [8], RM211 and RM53 on chromosome 2 [47], R1962 and RM225 on chromosome 6 [3],[47], RM201 on chromosome 9 [50], R2174 and R1629 on chromosome 10 [3], and C732 and R2672 on chromosome 12 [3],[51].
QTLs for HI
Three QTLs for HI were mapped and collectively explained 20.5% of the total phenotypic variation. Minghui 63, with a lower HI relative to Zhenshan 97, provided the two alleles at qHI1 and qHI11, which explained 17.1% of the total variation. The QTL qHI11 was also detected in the environment e4, and it had a significant interaction with the environment.
QTLs for Yield
Two QTLs were identified for Yield, and they jointly explained 11.2% of the total phenotypic variation. The QTL qYield2 with the low-score parent Zhenshan 97 allele had a large effect and contributed 8.1% of the total variation. The other QTL, which had the Minghui 63 allele, contributed 3.1%. The QTL qYield2 was mapped in three individual environments. The QTL qYield2 were detected in three environments across two years.
QTLs for PUP
Three QTLs for PUP were identified and explained 13.6% of the total phenotypic variation. Among these three QTLs, the two alleles at qPUP7 and qPUP10 were from Minghui 63, which had a higher PUP than Zhenshan 97. The QTLs qPUP1 and qPUP10 were identified in multiple environments.
QTLs for PHI
Four additive QTLs for PHI were found, and they collectively accounted for 42.8% of the total phenotypic variation. The contribution of each QTL ranged from 2.6% to 20.0%. Minghui 63 provided the alleles at two QTLs, qPHI1 and qPHI11. The alleles for increasing PHI at the other two QTLs, qPHI2 and qPHI6, were from Zhenshan 97. The qPHI1 with the Minghui 63 allele explained 15.8% of the total variation, whereas the qPHI2 with the Zhenshan 97 allele contributed 20.0%. A significant interaction was detected only between qPHI11 and e4. The two QTLs on chromosomes 1 and 6 were found in two environments simultaneously.
QTL for gPUE
Only one QTL, qgPUE4, was identified for gPUE. It was located in the region BIN680–BIN681 on chromosome 4 and explained 1.4% of the total phenotypic variation.
QTLs for strPUE
For strPUE, three QTLs were verified on chromosomes 1 and 2, and together they accounted for 15.1% of the phenotypic variation. The Minghui alleles at two QTLs (qstrPUE1-1 and qstrPUE1-2) and the Zhenshan97 allele at qstrPUE2 increased strPUE. The two QTLs (qstrPUE1-2 and qstrPUE2) were detected in three environments across two years.
QTL for PUEb
Only one QTL (qPUEb2) controlling PUEb on chromosome 2 was detected, and it explained 6.4% of the phenotypic variation. This QTL was only found in a single environment.
QTLs for PUEg
Five QTLs were detected for PUEg, collectively accounting for 32.6% of the phenotypic variation. The QTL (qPUEg2) on chromosome 2 had large additive effect, accounting for 13.4% of the phenotypic variation. The alleles for increasing PUEg came from both Minghui 63 at three QTLs (qPUEg1, qPUEg11, and qPUEg12) and Zhenshan 97 at two QTLs (qPUEg2 and qPUEg6). Two QTLs (qPUEg11 and qPUEg12) had significant interactions with the environment. Three QTLs were located in individual environments simultaneously.
QTLs for PT
For PT, three QTLs (qPT2, qPT5, and qPT8) were detected on chromosomes 2, 5, and 8, accounting for 14.5% of the phenotypic variation (Table 4). At these three loci, the alleles from Zhenshan 97 increased the trait. All these QTLs were detected in individual environments.
QTLs for PTE
Six QTLs for PTE were detected on chromosomes 1, 2, 5, 8, and 12, and they accounted for 34.1% of the phenotypic variation. The Minghui 63 alleles increased PTE at the three QTLs (qPTE1-1, qPTE1-2, and qPTE12), and the Zhenshan 97 alleles increased the trait score for the remaining QTLs (qPTE2, qPTE5, and qPTE8). The QTL qPTE2 had a significant interaction with the environment. Among the six QTLs, four were identified in individual environments.
Co-location and cluster of QTLs
Thirty-one QTLs were mapped on the same location or clustered in 12 intervals, respectively (Table 5). There were three regions on chromosome 1. The Minghui 63 alleles on two regions (BIN59–BIN61 and BIN143–BIN161) increased the phenotypic score, whereas the region BIN31–BIN47 covered favorable alleles from the two parents. There were three regions on chromosome 2 in which alleles from Zhenshan 97 increased the phenotypic score. The other six regions were located on chromosomes 5, 7, 8, 10, 11, and 12. Among the six regions, Zhenshan 97 contributed alleles for increasing phenotypic score to two regions (BIN708–BIN710 and BIN1130–BIN1132), and Minghui 63 provided alleles in the remaining four regions. Seven of the 12 intervals contained clustered QTLs for yield and PUE traits simultaneously.
Detection of epistatic interactions
Twenty-four digenic interactions were detected for eight of the eleven traits (Table 6). Each interaction explained 0.4% to 10.2% of the phenotypic variation. Eighteen of the twenty-four interactions did not involve loci with additive effects. Of the six interactions involving additive QTLs, three occurred between two additive QTLs. The first occurred between the two main QTLs for PUP, qPUP1 and qPUP7. The second additive interaction was between QTLs for PHI and existed between the regions BIN59–BIN60 and BIN1392–BIN1393 for PHI; QTLs for PHI, strPUE, and HI (qPHI1, qstrPUE1-1, and qHI1, respectively) were located on the former region. The third interaction occurred between BIN160–BIN161 and BIN310–BIN311 for QTLs of PTE; the former region contained a QTL for PTE (qPTE1-2), and the latter contained the QTLs qPTE2 and qPHI2. Three significant interactions occurred between e4 and QTLs for BIOM, PHI, and PTE (normal P in 2009), and each aae interaction explained 0.5%, 0.5%, and 0.8% of the phenotypic variation, respectively.
Discussion
Interrelationships among PUE traits
Two PUE traits (gPUE and strPUE) may be considered as components of P use efficiency [32]. There were positive correlations among PUEb, PUEg, and gPUE, and among PUEb, PUEg, and strPUE (Table 3 and Figure 1). However, we did not find any regions simultaneously containing QTLs for PUEb, PUEg, and gPUE, or PUEb, PUEg, and strPUE. Moreover, we did not find any co-locations of QTLs for PUEg and gPUE or for PUEb and strPUE. Therefore, P use efficiencies based on the P stored in a specific part of plant (such as gPUE and strPUE) is distinct from those based on the total P in an entire plant (such as PUEg and PUEb). No correlation occurred between strPUE and gPUE, and the co-location of QTLs for the two traits was not observed either. This suggests that strPUE and gPUE are independent from each other. Therefore, our results showed that it might be feasible to improve gPUE and strPUE independently. The majority of accumulated P was distributed in rice grains, and improving gPUE (such as decreasing P concentration in grains) may therefore be a suitable option for reducing plant P demand.
A genotype with tolerance to P deficiency is desirable, with simultaneously high PUP and high PUE [1],[3]. Negative correlations between PUP and PUEb, PUEg, gPUE, and strPUE were observed (Table 3, Figure 1). Similar correlations have been previously reported in rice [3] and wheat [15],[18],[33]. Manske et al. [40] showed that wheat PUEg was negatively correlated with pre-anthesis P accumulation, but positively correlated with post-anthesis P accumulation. In our study, PUEg was negatively correlated with pre-anthesis P accumulation under low P conditions (r = −0.27** in 2008, −0.29** in 2009, ** indicate significance at P < 0.01) and not correlated with post-anthesis P accumulation under normal P condition. However, gPUE was negatively correlated with post-anthesis P accumulation (r = −0.41** under low P in 2008, −0.32** under low P and −0.38** under normal P in 2009). Therefore, the contributions of P accumulations to PUE appear to be different at various growth stages, depending on crop and P level.
The QTLs for PUP did not share any regions with those for the four PUE traits based on grain yield and biomass (PUEb, PUEg, gPUE, and strPUE). Genetic association (close linkage and pleiotropy) and environmental effects are the main causes for the correlations between the traits [41]. Thus, the relationships between PUP and the four PUE traits may not be directly due to genetic association. In fact, P accumulation is involved in root characters, P absorption, and translocation and distributions among various parts of a plant, whereas PUE is associated with biomass production and yield formation, as well as with responses of a plant to P supply (Figure 1).
Relationships among yield and PUE traits
Seven regions simultaneously controlling yield and PUE traits (Table 5) were identified. These co-locations implied the close association between yield and PUE traits, which is also shown by the correlation analysis (Table 3, Figure 1). Generally, P accumulation and PUEg increased with yield and biomass. As expected, PUP, PUEg, and PUEb were individually positively correlated with yield. Regardless of these correlations, this study did not identify any common regions simultaneously shared by QTLs for PUP, PUEg/PUEb, and Yield. The QTLs for Yield and PUEg shared the interval BIN302–BIN303. This is consistent with the fact that high grain yield often results in high PUEg when P content is stable. We found co-locations for PUEg and PTE and for PUEg and PT, suggesting close relationships of PUEg with PTE and PT. The translocation of P from stems to grains is associated with grain P accumulation and PHI during grain filling [13]. Although Ogawa et al. [42] reported that carbohydrate accumulation is not directly associated with P accumulation in rice grains, increased P translocation to younger panicles may facilitate the grain filling and lead to high grain yield, which is advantageous to PUEg.
In this study, PHI was associated with HI and P contents in grains and straw. The region BIN1392–BIN1402 on chromosome 11 contained four QTLs affecting PHI, PUEg, HI, and Yield. This co-location was consistent with the correlations found in this study and in previous studies [14],[33]. In contrast, two regions (BIN59–BIN60 and BIN1392–BIN1399) contained QTLs for HI and PHI (Table 5 and Figure 2). However, the QTLs for PHI were different than those for HI, which suggests a relative independence of the two traits.
Grain yield was taken into account when HI and PUEg were calculated. Higher grain yield may indicate both higher HI and higher PUEg when the P accumulation in an entire plant is the same, as supported by the high correlation between the two traits (Table 3). The two regions on chromosomes 2 and 11 were simultaneously shared by QTLs for HI and PUEg (Table 5). Both the co-location of these QTLs and the correlation between HI and PUEg suggest that PUEg depends on HI to some degree. However, the two traits had specific QTLs that were not common, implying different genetic controls. From the viewpoint of crop physiology, HI is positively associated with grain yield and total biomass, whereas PUEg depends on grain yield, in addition to the P content of an entire plant (Table 3, Figure 1).
Plant P accumulation increases biomass production to a certain extent and vice versa [3],[15],[18], as confirmed by the correlations found in this study (Table 3). Three regions were concurrently shared by QTLs for BIOM and PUP (Table 5). Notably, at least two QTLs for BIOM were not shared by PUP, and no correlation was observed between growth duration and PUP (data not shown), suggesting that the processes for biomass production and P accumulation in rice are not the same. With respect to physiology, biomass production is related to photosynthetic capacity and respiration, and P accumulation is often affected by P absorption and translocation. Moreover, two QTLs for PUEb and BIOM were tightly linked in the region BIN244–BIN254 on chromosome 2, and two regions (BIN294–BIN303 and BIN1392–BIN1402) simultaneously affected PUEg and Yield (Table 5 and Figure 2). This suggests that Yield and PUEg (and BIOM and PUEb) could be concurrently improved by exploring these regions.
Epistatic and environmental effects for PUE traits
Epistasis is the interaction of genes at different loci, and it plays an important role in the formation of complex traits [43],[44]. A dozen pairs of epistatic interactions were detected for PUE traits in this study. Taking PUP as an example, three QTLs explained 13.6% of the phenotypic variation, and three pairs of interactions explained 9.0% of the variation (Tables 4 and 6). This implies that both additive QTLs and epistatic interactions could make substantial contributions to PUE traits. A relatively large proportion of QTLs (33%, 12 of 36) had both individual effects and epistatic interactions (Tables 4 and 6), indicating that interaction is common for yield and PUE traits. Hu et al. [45] and Li et al. [8] have reported similar epistatic interactions.
The development of complex traits integrates genetic and environmental effects. Seven of the 36 additive QTLs (Table 4) and the three epistatic interactions (Table 6) identified in this study interacted significantly with the environment. The seven QTLs with ae effects exhibited various responses to the environment. Several interactions of QTLs with the environment and epistasis with the environment were up-regulated by the particular environment. For example, three additive QTLs (qBIOM7, qHI11, and qPHI11, Table 4) and three pairs of epistatic interactions (Table 6) displayed positive ae and aae effects with the environment e4 (normal P in 2009), suggesting that the QTLs and epistatic interactions were significantly strengthened by the application of P fertilizer.
In contrast, several interactions were down-regulated by the environment. For example, two QTLs controlling PUEg (qPUEg11, qPUEg12) were down-regulated by e2 (low P in 2009), and two QTLs (qBIOM2 and qPTE2) were down-regulated by e4 (normal P in 2009). The down-regulations of qPUEg11 and qPUEg12 by low P and the down-regulation of qBIOM2 by the normal P application appeared in contrast to the expected behavior, as low P application often results in high PUEg and low BIOM (Table 1). However, for qPTE2, the down-regulation by the normal P application was consistent with the reduction of PTE under normal P. Therefore, our results suggest that both QTL and epistatic interactions are differentially expressed in response to the environment.
Comparisons of QTLs across genetic backgrounds
In previous studies, several populations have been used to detect QTLs associated with PUE in rice, with more attention focused on P deficiency tolerance and relative agronomic traits [3],[8],[17],[45],[46]. According to the physical positions in the SNP (Figure 2) and RFLP/SSR maps [28], the QTL qPUP10 for PUP may share a similar region with a QTL for PUP on chromosome 10 found by Wissuwa et al. [3], which was flanked by the marker R1629. Similarly, the QTL qPUP1 (BIN46–BIN47) was very close to the marker RM259 on chromosome 1 for root traits, which was found by Li et al. [8]. A QTL interaction linked to PUP on chromosome 2 was located on an interval (BIN241–BIN242) that was tightly linked to marker RM211, which was close to a QTL for root number [47]. These results were consistent with previous studies showing that root traits play an important role in P uptake and tolerance to P deficiency [48],[49].
Further increases in grain yield under P deficiency would most likely result from increases in PUP and PHI [33]. The interval BIN1290–BIN1291 on chromosome 9, which was involved in an interaction for PHI, had a similar location to RM201; moreover, Mao et al. [50] identified a QTL for 1000-grain weight on the similar interval (Figure 2). The QTL, qPHI6 for PHI seems to share a similar region with the QTLs near R1962 on chromosome 6 for tiller number and PUP under P deficiency detected by Wissuwa et al. [3]. This suggests that the QTLs and the interval may be involved in partitioning more P to grains under P deficient conditions.
The interval BIN1549–BIN1550, which was involved in an epistatic interaction for PTE on chromosome 12, was tightly linked to R2672, which was close to two QTLs for P-deficiency tolerance [3] and for amount of stem non-structural carbohydrates per spikelet at heading [51]. Thus, considering the traits linked to and controlled by the same interval, the epistatic effect between BIN1299–BIN1300 and BIN1549–BIN1550 may contribute to the relocations of P and carbohydrates from stems to grains during grain filling.
Conclusions
In this study, most of the QTLs for the eight PUE traits were different from those for yield traits, indicating different genetic mechanisms underlying high PUE and high grain yield. However, seven regions were shared by yield and PUE traits; therefore, grain yield and PUE traits may be simultaneously improved.
Four QTLs (qPHI1 and qPHI2 for PHI, qPUEg2 for PUEg, and qPTE8 for PTE) had strong additive effects but no environmental effects (Table 4). Additionally, three QTLs (qPUP1, qPUP10, and qPHI6) were simultaneously found under different genetic backgrounds. These QTLs are promising for improving rice PUE in future breeding research.
This study documented that 12 QTLs and many loci were involved in epistatic interactions, which played substantial roles in determining PUE traits. The observed interactions provide an approach to reveal the genetic networks affecting PUE traits.
Abbreviations
- BIOM:
-
Biomass
- gPUE:
-
Grain P use efficiency based on P accumulation in grains
- HI:
-
Harvest index
- PHI:
-
P harvest index
- PT:
-
P translocation
- PTE:
-
P translocation efficiency
- PUEb:
-
P use efficiency for biomass
- PUEg:
-
P use efficiency for grain yield based on P accumulation in the whole plant
- PUP:
-
Total P uptake in the whole plant
- QTLs:
-
Quantitative trait loci
- strPUE:
-
Straw P use efficiency based on P accumulation in straw
References
Panigrahy M, Rao DN, Sarla N: Molecular mechanisms in response to phosphate starvation in rice. Biotechnol Adv. 2009, 27: 389-397. 10.1016/j.biotechadv.2009.02.006.
Vance CP: Symbiotic nitrogen fixation and phosphorus acquisition, plant nutrition in world of declining renewable resources. Plant Physiol. 2001, 127: 390-397. 10.1104/pp.010331.
Wissuwa M, Yano M, Ae N: Mapping of QTLs for phosphorus-deficiency tolerance in rice (Oryza sativa L.). Theor Appl Genet. 1998, 97: 777-783. 10.1007/s001220050955.
Ismail AM, Heuer S, Thomson MJ, Wissuwa M: Genetic and genomic approaches to develop rice germplasm for problem soils. Plant Mol Biol. 2007, 65: 547-570. 10.1007/s11103-007-9215-2.
Ramaekers L, Remans R, Rao IM, Blair MW, Vanderleyden J: Strategies for improving phosphorus acquisition efficiency of crop plants. Field Crop Res. 2010, 117: 169-176. 10.1016/j.fcr.2010.03.001.
Hammond JP, Broadley MR, White PJ: Genetic responses to phosphorus deficiency. Ann Bot. 2004, 94: 323-332. 10.1093/aob/mch156.
Bucher M: Functional biology of plant phosphate uptake at root and mycorrhiza interfaces. New Phytol. 2007, 173: 11-26. 10.1111/j.1469-8137.2006.01935.x.
Li JZ, Xie Y, Dai AY, Liu LF, Li ZC: Root and shoot traits responses to phosphorus deficiency and QTL analysis at seedling stage using introgression lines of rice. J Genet Genomics. 2009, 36: 173-183. 10.1016/S1673-8527(08)60104-6.
Rausch C, Bucher M: Molecular mechanisms of phosphate transport in plants. Planta. 2002, 216: 23-37. 10.1007/s00425-002-0921-3.
Rengel Z: Genetic control of root exudation. Plant Soil. 2002, 245: 59-70. 10.1023/A:1020646011229.
Heuer S, Lu XC, Chin JH, Tanaka JP, Kanamori H, Matsumoto T, De Leon T, Ulat VJ, Ismail AM, Yano M, Wissuwa M: Comparative sequence analyses of the major quantitative trait locus phosphorus uptake 1 (Pup1) reveal a complex genetic structure. Plant Biotechnol J. 2009, 7: 456-471. 10.1111/j.1467-7652.2009.00415.x.
Wissuwa M, Ae N: Genotypic variation for tolerance to phosphorus deficiency in rice and the potential for its exploitation in rice improvement. Plant Breed. 2001, 120: 43-48. 10.1046/j.1439-0523.2001.00561.x.
Dordas C: Dry matter, nitrogen and phosphorus accumulation, partitioning and remobilization as affected by N and P fertilization and source-sink relations. Eur J Agron. 2009, 30: 129-139. 10.1016/j.eja.2008.09.001.
Rose TJ, Pariasca-Tanaka J, Rose MT, Fukuta Y, Wissuwa M: Genotypic variation in grain phosphorus concentration, and opportunities to improve P-use efficiency in rice. Field Crop Res. 2010, 119: 154-160. 10.1016/j.fcr.2010.07.004.
Su JY, Xiao YM, Li M, Liu QY, Li B, Tong YP, Jia JZ, Li ZS: Mapping QTLs for phosphorus-deficiency tolerance at wheat seedling stage. Plant Soil. 2006, 2006 (281): 25-36. 10.1007/s11104-005-3771-5.
Paterson AH, Lander ES, Hewitt JD, Peterson S, Lincoln SE, Tanksley SD: Resolution of quantitative traits into Mendelian factors by using a complete linkage map of restriction fragment length polymorphisms. Nature. 1988, 335: 721-726. 10.1038/335721a0.
Ni JJ, Wu P, Senadhira D, Huang N: Mapping QTLs for phosphorus deficiency tolerance in rice (Oryza sativa L.). Theor Appl Genet. 1998, 97: 1361-1369. 10.1007/s001220051030.
Su JY, Zheng Q, Li HW, Li B, Jing RL, Tong YP, Li ZS: Detection of QTLs for phosphorus use efficiency in relation to agronomic performance of wheat grown under phosphorus sufficient and limited conditions. Plant Sci. 2009, 176: 824-836. 10.1016/j.plantsci.2009.03.006.
Chen JY, Xu L, Cai YL, Xu J: Identification of QTLs for phosphorus utilization efficiency in maize (Zea mays L.) across P levels. Euphytica. 2009, 167: 245-252. 10.1007/s10681-009-9883-x.
Zhu JM, Kaeppler SM, Lynch JP: Mapping of QTLs for lateral root branching and length in maize (Zea mays L.) under differential phosphorus supply. Theor Appl Genet. 2005, 111: 688-695. 10.1007/s00122-005-2051-3.
Li YD, Wang YJ, Tong YP, Gao JG, Zhang JS, Chen SY: QTL mapping of phosphorus deficiency tolerance in soybean (Glycine max L. Merr.). Euphytica. 2005, 142: 137-142. 10.1007/s10681-005-1192-4.
Zhang D, Cheng H, Geng LY, Kan GZ, Cui SY, Meng QC, Gai JY, Yu DY: Detection of quantitative trait loci for phosphorus deficiency tolerance at soybean seedling stage. Euphytica. 2009, 167: 313-322. 10.1007/s10681-009-9880-0.
Chaubey CN, Senadhira D, Gregorio GB: Genetic analysis of tolerance for phosphorous deficiency in rice (Oryza sativa L.). Theor Appl Genet. 1994, 89: 313-317.
Guo ZH, Ding P, He LY, Xu CG: Genetic analysis of agricultural traits in rice related to phosphorus efficiency. Acta Genet Sin. 2006, 33: 634-641. 10.1016/S0379-4172(06)60093-0.
Li ZK, Yu SB, Lafitte HR, Huang N, Courtois B, Hittalmani S, Vijayakumar CHM, Liu GF, Wang GC, Shashidhar HE, Zhuang JY, Zheng KL, Singh VP, Sidhu JS, Srivantaneeyakul S, Khush GS: QTL × environment interactions in rice. I. Heading date and plant height. Theor Appl Genet. 2003, 108: 141-153. 10.1007/s00122-003-1401-2.
Li M, Guo XH, Zhang M, Wang XP, Zhang GD, Tian YC, Wang ZL: Mapping QTLs for grain yield and yield components under high and low phosphorus treatments in maize (Zea mays L.). Plant Sci. 2010, 178: 454-462. 10.1016/j.plantsci.2010.02.019.
Bray EA: Plant responses to water deficit. Trends Plant Sci. 1997, 2: 48-54. 10.1016/S1360-1385(97)82562-9.
Yu HH, Xie WB, Wang J, Xing YZ, Xu CG, Li XH, Xiao JH, Zhang QF: Gains in QTL detection Using an ultra-high density SNP map based on population sequencing relative to traditional RFLP/SSR markers. PLoS One 2011, 6:e17595.,
Wang J, Yu HH, Xie WB, Xing YZ, Yu SB, Xu CG, Li XH, Xiao JH, Zhang QF: A global analysis of QTLs for expression variations in rice shoots at the early seedling stage. Plant J. 2010, 63: 1063-1074. 10.1111/j.1365-313X.2010.04303.x.
Hyten DL, Song QJ, Choi IY, Yoon MS, Specht JE, Matukumalli LK, Nelson RL, Randy C, Shoemaker RC, Young ND, Cregan PB: High-throughput genotyping with the GoldenGate assay in the complex genome of soybean. Theor Appl Genet. 2008, 116: 945-952. 10.1007/s00122-008-0726-2.
Yan JB, Yang XH, Shah T, Sánchez-Villeda H, Li JS, Warburton ML, Zhou Y, Crouch JH, Xu YB: High-throughput SNP genotyping with the GoldenGate assay in maize. Mol Breed. 2010, 25: 441-451. 10.1007/s11032-009-9343-2.
Rose TJ, Wissuwa M: Rethinking internal phosphorus utilization efficiency: a new approach is needed to improve PUE in grain crops. Adv Agron. 2012, 116: 185-217. 10.1016/B978-0-12-394277-7.00005-1.
Jones GPD, Blair GJ, Jessop RSJ: Phosphorus efficiency in wheat - A useful selection criterion?. Field Crop Res. 1989, 21: 257-264. 10.1016/0378-4290(89)90007-5.
Xing YZ, Tan YF, Hua JP, Sun XL, Xu CG, Zhang Q: Characterization of the main effects, epistatic effects and their environmental interactions of QTLs on the genetic basis of yield traits in rice. Theor Appl Genet 2002, 105: 248–257. 33
Murphy J, Riley JP: A modified single solution method for the determination of phosphate in natural waters. Anal Chim Acta. 1962, 27: 31-36. 10.1016/S0003-2670(00)88444-5.
Knapp SJ, Stroup WW, Ross WM: Exact confidence intervals for heritability on a progeny mean basis. Crop Sci. 1985, 25: 192-194. 10.2135/cropsci1985.0011183X002500010046x.
Xie WB, Feng Q, Yu HH, Huang XH, Zhao Q, Xing YZ, Yu SB, Han B, Zhang QF: Parent-independent genotyping for constructing an ultra high-density linkage map based on population sequencing. Proc Natl Acad Sci USA. 2010, 107: 10578-10583. 10.1073/pnas.1005931107.
Wang DL, Zhu J, Li ZK, Paterson AH: Mapping QTLs with epistatic effects and QTL × environment interactions by mixed linear model approaches. Theor Appl Genet. 1999, 99: 1255-1264. 10.1007/s001220051331.
McCouch SR, Cho YG, Yano M, Paul E, Blinstrub M: Report on QTL nomenclature. Rice Genet Newsl. 1997, 14: 11-13.
Manske GGB, Ortiz-Monasterio JI, van Ginkel RM, Rajaram S, Vlek PLG: Phosphorus use efficiency in tall, semi-dwarf and dwarf near-isogenic lines of spring wheat. Euphytica. 2002, 125: 113-119. 10.1023/A:1015760600750.
Aastveit AH, Aastveit K: Effects of genotype-environment interactions on genetic correlations. Theor Appl Genet. 1993, 86: 1007-1013. 10.1007/BF00211054.
Ogawa M, Tanaka K, Kasai Z: Accumulation of phosphorus, magnesium and potassium in developing rice grains: followed by electron microprobe X-ray analysis focusing on the aleurone layer. Plant Cell Physiol. 1979, 20: 19-27.
Carlborg Ö, Haley CS: Epistasis: too often neglected in complex trait studies?. Nat Rev Genet. 2004, 5: 618-625. 10.1038/nrg1407.
Wu XS, Wang ZH, Chang XP, Jing RL: Genetic dissection of the developmental behaviours of plant height in wheat under diverse water regimes. J Exp Bot. 2010, 61: 2923-2937. 10.1093/jxb/erq117.
Hu B, Wu P, Liao CY, Zhang WP, Ni JJ: QTLs and epistasis underlying activity of acid phosphatase under phosphorus sufficient and deficient conditions in rice (Oryza sativa L.). Plant Soil. 2001, 230: 99-105. 10.1023/A:1004809525119.
Shimizu A, Yanagihara S, Kawasaki S, Ikehashi H: Phosphorus deficiency-induced root elongation and its QTL in rice (Oryza sativa L.). Theor Appl Genet. 2004, 109: 1361-1368. 10.1007/s00122-004-1751-4.
Cui KH, Huang JL, Xing YZ, Yu S, Xu CG, Peng SB: Mapping QTLs for seedling characteristics under different water supply conditions in rice (Oryza sativa). Physiol Plantarum. 2008, 132: 53-68.
Itoh S, Barber SA: A numerical solution of whole plant nutrient uptake for soil-root systems with root hairs. Plant Soil. 1983, 70: 403-413. 10.1007/BF02374895.
Wissuwa M: Combining a modelling with a genetic approach in establishing associations between genetic and physiological effects in relation to phosphorus uptake. Plant Soil. 2005, 269: 57-68. 10.1007/s11104-004-2026-1.
Mao DH, Liu TM, Xu CG, Li XH, Xing YZ: Epistasis and complementary gene action adequately account for the genetic bases of transgressive segregation of kilo-grain weight in rice. Euphytica. 2011, 180: 261-271. 10.1007/s10681-011-0395-0.
Nagata K, Shimizu H, Terao T: Quantitative trait loci for nonstructural carbohydrate accumulation in leaf sheaths and culms of rice (Oryza sativa L.) and their effects on grain filling. Breed Sci. 2002, 52: 275-283. 10.1270/jsbbs.52.275.
Acknowledgements
We thank Professors Xing Yongzhong and Yu Sibin, and the National Key Laboratory of Crop Genetic Improvement, Huazhong Agricultural University, Wuhan, 430070, for providing seeds and the SNP marker data for the recombinant inbred lines used in the study. This study was supported by the National Basic Research Program of China (2005CB120905) and the National Key Technology R&D Program (2013BAD07B10) of the Ministry of Science and Technology.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KC, KW, and JH designed the experiments. KW, GL, JP, and KC performed the field experiments, P concentration determinations, and data analyses. WX and HY constructed the SNP map; LN and JH provided field management; KW and KC wrote the manuscript; FS and SP interpreted the data and revised the manuscript. All authors have read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
Cite this article
Wang, K., Cui, K., Liu, G. et al. Identification of quantitative trait loci for phosphorus use efficiency traits in rice using a high density SNP map. BMC Genet 15, 155 (2014). https://doi.org/10.1186/s12863-014-0155-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-014-0155-y