
Abstract
Background
Identification of multi-markers is one of the most challenging issues in personalized medicine era. Nowadays, many different types of omics data are generated from the same subject. Although many methods endeavor to identify candidate markers, for each type of omics data, few or none can facilitate such identification.
Results
It is well known that microRNAs affect phenotypes only indirectly, through regulating mRNA expression and/or protein translation. Toward addressing this issue, we suggest a hierarchical structured component analysis of microRNA-mRNA integration (“HisCoM-mimi”) model that accounts for this biological relationship, to efficiently study and identify such integrated markers. In simulation studies, HisCoM-mimi showed the better performance than the other three methods. Also, in real data analysis, HisCoM-mimi successfully identified more gives more informative miRNA-mRNA integration sets relationships for pancreatic ductal adenocarcinoma (PDAC) diagnosis, compared to the other methods.
Conclusion
As exemplified by an application to pancreatic cancer data, our proposed model effectively identified integrated miRNA/target mRNA pairs as markers for early diagnosis, providing a much broader biological interpretation.
Similar content being viewed by others

Background
Presently, numerous types of “omics” data are generated by many accurate and cost-effective methods. For instance, next-generation sequencing (NGS) technology is used to find DNA or RNA variations, bisulfite sequencing is used to find DNA-methylated variants, and multiple reaction monitoring (MRM) is applied to measure protein abundances [1,2,3]. These efficient omics data platforms allow researchers to use multi-omics data, obtained from the same subjects, for analyzing huge numbers of variants. As a result, efficient multi-omics data analysis is becoming more important in integrating large-scale data sets, making it possible to interpret fundamental biological systems [4].
MicroRNAs (miRNAs) are noncoding RNAs having a length less than 25 base pairs, regulating the expression of specific genes by mRNA degradation or blocking translation by binding to the 3′ regions of their “target” mRNAs. Many recent studies have now implicated miRNAs in the pathogenesis of cancer, including triggering cancer initiation and progression. MiRNAs have been shown to have tissue-specific and disease-specific expression patterns [5,6,7,8]. Intensive investigation is now underway for using applying miRNAs’ inhibitory information to mRNAs. For example, Nam et al. developed “miRNA and mRNA integrated analysis” (MMIA) to examine biological functions of miRNA expression [9]. Moreover, Buffa et al. used pathway information to independently validate miRNAs significant for breast cancer [10], while Cho et al. performed network analysis, and hierarchical clustering, to find biological “signatures” of interstitial lung diseases [11]. Most miRNA and mRNA integration analyses focus on first identifying miRNAs significantly associated with the phenotype of interest, and then experimentally validating those miRNAs’ phenotype involvement by inhibiting or ectopically overregulating their expression [9,10,11]. Although these approaches are effective at validating significant miRNAs, they do not provide information on how they regulate expression of their target mRNAs, as relevant to the pathway level.
In this work, we propose a structured component-based analysis, for integrating omics data for identifying multiple accurate biomarkers. It is well known that miRNAs affect phenotypes indirectly, by regulating mRNA expression or protein translation [8]. Herein, we propose hierarchical structured component analysis of miRNA-mRNA integration (HisCoM-mimi) analysis, which models biological relationships as structured components, to efficiently yield integrated markers. Our proposed model is based on generalized structured component analysis (GSCA), which tests hypothesized relationships between observed and latent variables [12]. GSCA is a component-based method whereby each component represents a latent variable. Extending GSCA, we previously developed Pathway-based approach using hierarchical components of collapsed rare variants (PHARAOH) [13]. PHARAOH uses a hierarchial structure of rare variants, genes, and pathways. The advantage of such hierarchical structural component models is their generation of (unobservable) latent variables, such as genes and pathways, which are inferred by observed variables, such as rare variants. Using latent variables, we can collapse unstructured data into a structured form, providing less ambiguous biological explanations of the results. In this current work, mRNAs, inhibited by miRNAs, can be merged into latent variables.
Accordingly, our proposed HisCoM-mimi model can efficiently account for biological relationships between miRNA and mRNA, in the structured component, and effectively provide integrated (e.g., miRNA-to-target-mRNA) markers. As an illustration, we tried HisCoM-mimi for identifying biomarkers for the early diagnosis of pancreatic cancer (PC). Note that PC is one of the most fatal diseases in the world, having a mere 8% five-year survival rate in the USA and a 9.4% survival rate in the Republic of Korea [14,15,16]. In particular, the tumor heterogeneity in PC patients’ tumors makes early diagnosis harder than cancers of most other organs [17]. To adjust for heterogeneity among tumor cells, we need a more robust and complex statistical model which can interpret and integrate several causes of cancer altogether. Although many bioinformatics research studies have been performed to find diagnostic markers for PC, to date, no clinically approved prognostic markers exist [18].
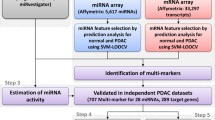
Here, we applied HisCoM-mimi to computationally identify diagnostic markers of pancreatic ductal adenocarcinoma (PDAC), the most common type of PC. By applying the HisCoM-mimi approach to miRNA and mRNA microarray data from PDAC patients, at Seoul National University Hospital (SNUH), we identified numerous cognate miRNA-mRNA partners, as markers for diagnosis of PDAC. Finally, our HisCoM-mimi provided integrated marker sets, with more biological and intuitive interpretation, than other existing methods.
Methods
Pancreatic ductal adenocarcinoma (PDAC) samples
Between the years 2009 and 2012, 200 pancreatic ductal adenocarcinoma (PDAC) samples were collected by the Department of Hepatobiliary and Pancreas Surgery of Seoul National University Hospital. The study protocol was approved by the Institutional Review Board of Seoul National University Hospital (IRB H-0901-010-267) and written, informed consent was obtained from each patient or legally authorized representative.
Of the 200 tumors, 96 were excluded because of RNA degradation or insufficient RNA content, leaving 104 samples valid for microarray analysis. After quality control, 97 PDAC samples remained for microarray assessment. The PDAC patients’ average age was 64.3 years (standard deviation (SD): 9.7). Twenty-nine patients were male, and 31 female. For the normal groups, 17 benign pancreatic tissues were used. Subsequently, we built and implemented our mini model, using the 97 PDAC and 17 normal tissues, respectively.
HisCoM-mimi model
To perform the integration analysis of miRNA and mRNA data, we developed and implemented our HisCoM-mimi approach. This model analyzes multiple subnetworks simultaneously, with specific regard to inverse correlations between mRNA and miRNA. Figure 1 shows the flowchart of the method. First, for a given miRNA, a miRNA-mRNA subnetwork, consisting of one miRNA and multiple potential target mRNAs, is constructed if the following two conditions are satisfied: (i) the mRNAs are reported as target of the miRNA by TargetScan 7.1 (targetscan.org) [19], and the negative correlation coefficients between the mRNA and miRNAs are significant (p-value < 0.05). Second, for all entities deemed significant, we derived our hierarchical structural component model by using all miRNA-mRNA subnetworks.

Flow chart for analyzing mRNA-miRNA integration
As shown in Fig. 2, there are three structures to consider: miRNA-mRNA structure, miRNA integration latent structure, and phenotype-latent structure. Each structure can be represented as a generalized linear model, similar to PHARAOH [13].

Network Diagram for HisCoM-mimi model
miRNA-mRNA structure
Equation (1) shows how to obtain mRNA expression before inhibition by miRNA, subscript i means i th individual, x ijk represents the mRNA expression of the kth gene related with j th miRNA, z j the j th miRNA expression, γ jk the inhibition coefficient for the j th miRNA for the k th gene, and G j is the number of inhibited mRNAs by the j th miRNA. By estimating the coefficients γ jk , mRNA expression after removing the inhibition effect of miRNA can be obtained.
miRNA latent structure
The miRNA latent variable is defined in Eq. (2). The miRNA latent variable is built by linearly combining miRNA expression values. While γj0 denotes the direct effect of the miRNA on the phenotype. Then, the latent variable f ij represents the global effect of the miRNA’s activity through its inhibited mRNAs.
Phenotype-latent structure
Let the phenotype variable y i be a binary variable, distinguishing PDAC from normal tissues. Let π i be the probability of y i = 1 (PDAC). logit(π i ) is the logit link function, β j represents the effect of f ij on the phenotype, as interpreted as a log-odds ratio.
Fitting the HisCoM-mimi algorithm
To estimate the parameters for HisCoM-mimi, we adopted our previously developed PHARAOH algorithm [13], which is based on the alternating least squares algorithm for the penalized log-likelihood function, with ridge parameters. Then, the objective function to maximize is given as follows:
where p(y i ; γ i , δ) is the probability distribution for the phenotype of the ith individual. λ m and λ mm are ridge parameters for miRNA-mRNA pairs of interest, representing the integrated latent components.
To maximize the objective function, φ1, the iterative reweighted least squares (IRWLS) algorithm is used. Note that when using IRWLS, maximizing φ1 is equivalent to minimizing the object function φ2.
Comparative models
To compare the results of HisCoM-mimi with other methods, we considered several alternative regression-based methods.
Firstly, we considered the ordinary penalized logistic regression (LR) methods such as lasso or elastic-net (EN) [20, 21]. Equation 7 shows the LR model, where θ j and ρ k represent the effect of the jth miRNA and the kth mRNA, respectively. Equation 8 is the objective function to maximize for finding optimal parameters with the penalty function P α (θ, ρ). When lasso is used, P α (θ, ρ) =∑k ∣ ρ k ∣ + ∑j ∣ θ j ∣.
If EN is used, \( {P}_{\alpha}\left(\theta, \rho \right)=\alpha \left({\sum}_{\mathrm{k}}\left|{\rho}_k\right|+{\sum}_{\mathrm{j}}\left|{\theta}_j\right|\right)+\left(1-\alpha \right)\left({\sum}_{\mathrm{k}}{\rho}_k^2+{\sum}_j{\theta}_j^2\right) \). Lasso or EN can then select the miRNAs and/or mRNAs of interest. However, these methods cannot use group information. Thus, ordinarily penalized LR methods cannot adequately account for the biological structure of miRNA-mRNA.
Secondly, we considered LR with a group lasso penalty (GL) [22], which has the benefit of using group information among the miRNAs and mRNAs of interest. In our analysis, a group can be defined as a set of one miRNA and its corresponding inhibited target mRNAs. GL uses the same LR in (8) with a different penalty function \( P\left(\theta, \rho \right)={\sum}_{j=1}^J\sqrt{\theta_j^2+{\sum}_{k=1}^{G_j}\left|{\rho}_k\right|} \). Via this penalty function, miRNA integration set can be selected together. However, the GL approach does not easily provide p-values for each set of independent variables.
To fit the penalized LR models, we first performed 3-fold cross-validation to find the optimal tuning parameter, δ. after which we fitted the models with all the data sets.
Simulation study
To compare HisCoM-mimi to the other three methods, we performed simulation studies and computed type I errors and power, simulating data from the same miRNA and mRNA data structure in our pancreatic cancer dataset. That is, we selected miRNA and mRNA data from the pancreatic cancer dataset, and then generated phenotype data iteratively from the LR model. We then considered two simulation scenarios. Scenario 1 assumed that a true causal integration set contains two mRNAs, with the same effect size. Scenario 2 assumed that a true causal integration set contains five mRNAs, with the same effect size. For each scenario, we randomly selected one causal miRNA-mRNA subnetwork, and then randomly selected another 9 miRNA-mRNA subnetworks, for which the number of inhibited mRNAs was less than 10. The selected miRNA-mRNA subnetworks for Scenario 1 are summarized in Table 1 and for Scenario 2 are in Table 2.
For Scenario 1, we used miR-217 as a true causal miRNA. To generate phenotypes, we considered the following LR model.
where π is the probability of observing a disease (Y = 1), z1 represents the true causal miRNA expression, and x1 and x2 represent two causal mRNA expression values. For type I error evaluation, we assumed β miRNA = β1 = β2 = 0. For power comparison, we generated simulation data sets under the assumption that β miRNA = β1 = 0.2, 0.25, 0.3, 0.35. For the given 114 (97 PDAC and 17 normal tissues) values of (z1, x1, x2), from our pancreatic cancer dataset, we simulated 1000 datasets.
For Scenario 2, we assumed that a true causal integration set contains five mRNAs, with the same effect size. In our dataset, miR-381 was the only miRNA having five inhibited target mRNAs. To generate phenotypes, we considered the following LR model:
where x1, …, x5 represent five causal mRNA expression values. As in Scenario 1, we assumed β miRNA = β1 = β2 = β3 = β4 = β5 = 0, for type I error evaluation, and β miRNA = β1 = β2 = β3 = β4 = β5 = 0.2, 0.25, 0.3, 0.35, for power comparison. For the given 114 values of (z1, x1, x2, x3, x4, x5) from the pancreatic cancer dataset, 1000 simulation datasets were generated. We used the significance level α = 0.05 for HisCoM-mimi, as an false positive rate (FPR) criterion. For lasso, EN, and group-lasso, we selected a threshold T which provides a comparable FPR to the type I error 0.05. T was determined by calculating the FPR for simulation settings such that a miRNA-mRNA subnetwork is selected when β miRNA ≠ 0 and \( K\left(={\sum}_{\mathrm{l}=1}^{\mathrm{L}}I\left({\beta}_l\ne 0\right)\right) \) exceeded the threshold T. Here, L is the number of inhibited mRNAs for true causal miRNA for each scenario: L = 2 for Scenario 1, and L = 5 for Scenario 2.
Results
Simulation results
For our analyses, we first determined the false positive error rates (FPRs) of each method, and chose the threshold values of T to make each penalized method provide (hold) FPRs close to 0.05. In Scenario 1, the type I error rate of HisCoM-mimi was 0.048 when α = 0.05. The FPRs of lasso were 0.054, when T was 1, and that of EN was 0.064, when T was 1. Since type I error rates of lasso and EN were nearly 0.05 when T = 1, we set T = 1 to evaluate power of those two methods. The FPR of GL, when choosing a causal miRNA integration set, 0.064.
For Scenario 2, Table 3 shows the FPRs for lasso and EN, when varying the threshold T. For this result, we found that the type I error of lasso and EN were similar to 0.05, when T = 1 and 2, respectively. The type I error rate of HisCoM-mimi was 0.054. On the other hand, GL did not select a causal miRNA integration set at all, such that the type I error rate was 0. Secondly, we compared the powers of each method for Scenarios 1 and 2. Figure 3 shows bar plots of powers for scenario 1, where the x-axis shows the effect sizes (i.e., beta coefficients), and the y-axis shows the power. HisCoM-mimi showed the highest power, while EN was second, Lasso was third, and GL was last. The same tendency is shown in Fig. 4, for Scenario 2. Figure 5 shows that the differences of power between HisCoM-mimi and the others were much larger than those of Scenario 1. Consequently, GL could not find any significant miRNA-mRNA integration sets under Scenario 1, due to its GL’s penalty being too strict for many mRNAs, whose beta values were small.

Power comparison for scenario 1

Power comparison for scenario 2

Venn Diagram for number of detected miRNAs for each method
Constructing miRNA-mRNA subnetworks
To use human mRNA and miRNA probes, we first filtered out non-annotated mRNA probes and non-human miRNA probes. After filtering, there were 22,077 mRNA probes and 3391 miRNA probes. To construct miRNA-mRNA subnetworks, we checked predicted target mRNAs, for each miRNA, from TargetScan 7.1 (targetscan.org) [19, 23]. Among predicted targets, we only selected mRNAs having significant Pearson correlation coefficients with a specific miRNA. After filtering, there were 55 miRNAs, and 2411 edges connected with mRNAs.
Integration analysis for the PDAC data
Table 4 shows the top significant weights of miRNA-mRNA integrations derived from HisCoM-mimi. To perform multiple comparison, we used false discovery rate (FDR) q-values summarized in the 7th column [24]. We could only find 12 miRNAs having q-values below 0.05. Tables 5 and 6 show the lists of the selected markers by lasso and EN, respectively. Since lasso and EN select markers without any group information, they selected miRNA and mRNA markers independently. There were no miRNAs selected by lasso or EN directly, with lasso yielding only two significant mRNAs, both related to miR-326. Other mRNAs were independently selected from different miRNAs. Consequently, there were only 12 markers selected by lasso. For EN, 58 mRNAs were selected. Similar to the lasso result, there were no selected miRNAs, although four miRNAs (miR-206, miR-3064, miR-222, and miR-326) connected to more than three mRNAs. Figure 5 shows a Venn diagram of the number of miRNAs selected by each method. Each number represents the total number of detected miRNAs and one in the parenthesis does the number of detected miRNAs whose relationship with pancreatic cancer were reported. HisCoM-mimi selected larger number of unique miRNAs and the majority of them were already were reported.
For the lasso group only one miRNA (miR-32) and whose related two mRNA (COL1A2, and BGN) were selected. Although miR-32 is not reported as pancreatic cancer marker, there were some reports that miR-32 is related with other cancers [25, 26].
Table 7 summarizes miRNAs detected by HisCoM-mimi, lasso, EN, or GL. Previously, miR-93, miR-219, miR-141, miR-222, miR-203, miR-132, miR-96, and miR-206 were reported to be pancreatic cancer-related markers [27,28,29,30,31,32,33,34,35]. Although other miRNAs detected by HisCoM-mimi, lasso, EN, or GL have not been reported for pancreatic cancer relation, miR-532, miR-590, miR-133b, miR-326, miR-708, miR-3064, and miR-32 were reported to associate with other cancer types [25, 36,37,38,39,40,41,42].
Table 8 shows the cross-validation (CV) results for comparing prediction performance for marker-sets selected by HisCoM-mimi, Lasso, EN, and Group Lasso. The first column indicates methods used to construct prediction model and the second column does the method to select marker sets. The third column shows the area under the Receiver Operating Characteristic curve (AUC) results performed by leave-one-out cross validation (LOOCV). This setting is from the previous study of Kwon et al. [23]. The fourth column indicates the average AUC values performed by four-fold CV with a hundred iterations. Here, we used four-fold and eight-fold CV to balance the number of samples in CV datasets. The fifth column indicates the average AUC values performed by eight-fold CV with a hundred iterations. For all selected marker-sets, all prediction models built by HisCoM-mimi showed the best performances yielding AUC values higher than 0.9 except the marker-set selected by Group lasso in which the number of markers is less than five and one path coefficient exists.
Discussion and conclusion
In this paper, we proposed and developed a novel method, hierarchical structured component analysis of microRNA-mRNA integration (“HisCoM-mimi”), to construct a component model to identifying significantly integrated miRNA-target-mRNA cognate pairs. Since HisCoM-mimi could use subgroup information, it yelded more results, as related to phenotypes (e.g. cancer, metabolic syndrome, and etc.), than those of other existing methods that lack network information.
In simulation studies, we compared the performances of HisCoM-mimi, lasso, EN, and GL. From that comparison, HisCoM-mimi showed better performance than the other three methods. Controlling type I error, by HisCoM-mimi, was easier for controlling FPRs than other methods, because HisCoM-mimi uses permutation based p-values. In particular, HisCoM-mimi could identify miRNA-mRNA integration sets in a much more flexible way, due to better use of a standard multiple testing framework, as compared to the other methods. In real data analysis, HisCoM-mimi succesfully identified more miRNA-mRNA integration sets for pancreatic ductal adenocarcinoma (PDAC) diagnosis, compared to the other methods. Among 12 miRNAs, whose q-values were below 0.05 by HisCoM-mimi, 7 miRNAs were previously reported to associate with a panreatic cancer [27,28,29,30,31,32,33,34,35]. EN found two miRNAs (miR-222, and miR-206) [30, 34]. Among two miRNAs selected by lasso, only miR-222 was reported to associate with pancreatic cancer.
Although HisCoM-mimi worked well for the PDAC data sets, further biological verification of those results are needed. In future studies, we will perform additional simulation analyses to evaluate the performance of HisCoM-mimi, under numerous conditions. Furthermore, HisCoM-mimi can be extended in many ways, for other types of phenotypes, such as time to event. Second, it can be easily applied to other cancer studies to identify miRNA-mRNA integration sets for early diagnosis and prognosis. Third, it can be extended to combine other types of omics data such as genomics, epignomics, and proteomics data. It is now established that dysregulated miRNAs play substantial roles in a myriad of diseases [43]. We firmly believe that these methods for miRNA identification and their target transcripts could yield effective biomarkers and therapeutic targets, in addition to providing better understanding of disease mechanisms and etiology.
Abbreviations
- AUC:
-
Area under the receiver operating characteristic curve
- CV:
-
Cross-validation
- EN:
-
Elastic-net
- FPR:
-
False positive rate
- GL:
-
Group lasso
- GSCA:
-
Generalized structured component analysis
- HisCoM-mimi:
-
Hierarchical structured component analysis of microRNA-mRNA integration
- IRWLS:
-
Iterative reweighted least squares
- JJ:
-
Jin-Young Jang
- LR:
-
Logistic regression
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PHARAOH:
-
Pathway-based approach using hierarchical components of collapsed rare variants
- SC:
-
Sungkyoung Choi
- SL:
-
Sungyoung Lee
- SNUH:
-
Seoul National University Hospital
- TP:
-
Taesung Park
- YK:
-
Yongkang Kim
References
Reis-Filho JS. Next-generation sequencing. Breast Cancer Res. 2009;11(Suppl 3):S12.
Bock C, Reither S, Mikeska T, Paulsen M, Walter J, Lengauer T. BiQ analyzer: visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics. 2005;21(21):4067–8.
Anderson L, Hunter CL. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol Cell Proteomics. 2006;5(4):573–88.
Joyce AR, Palsson BO. The model organism as a system: integrating ‘omics’ data sets. Nat Rev Mol Cell Bio. 2006;7(3):198–210.
Farazi TA, Hoell JI, Morozov P, Tuschl T. MicroRNAs in human cancer. Adv Exp Med Biol. 2013;774:1–20.
Kang SM, Lee HJ. MicroRNAs in human lung cancer. Exp Biol Med. 2014;239(11):1505–13.
Navarro A, Monzo M. MicroRNAs in human embryonic and cancer stem cells. Yonsei Med J. 2010;51(5):622–32.
Negrini M, Ferracin M, Sabbioni S, Croce CM. MicroRNAs in human cancer: from research to therapy. J Cell Sci. 2007;120(11):1833–40.
Nam S, Li M, Choi K, Balch C, Kim S, Nephew KP. MicroRNA and mRNA integrated analysis (MMIA): a web tool for examining biological functions of microRNA expression. Nucleic Acids Res. 2009;37(Web Server issue):W356–62.
Buffa FM, Camps C, Winchester L, Snell CE, Gee HE, Sheldon H, Taylor M, Harris AL, Ragoussis J. microRNA-associated progression pathways and potential therapeutic targets identified by integrated mRNA and microRNA expression profiling in breast cancer. Cancer Res. 2011;71(17):5635–45.
Cho JH, Gelinas R, Wang K, Etheridge A, Piper MG, Batte K, Dakhallah D, Price J, Bornman D, Zhang S, et al. Systems biology of interstitial lung diseases: integration of mRNA and microRNA expression changes. BMC Med Genet. 2011;4:8.
Hwang HS, Takane Y. Generalized structured component analysis. Psychometrika. 2004;69(1):81–99.
Lee S, Choi S, Kim YJ, Kim BJ, Consortium T-G, Hwang H, Park T. Pathway-based approach using hierarchical components of collapsed rare variants. Bioinformatics. 2016;32(17):i586–94.
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. Ca Cancer J Clin. 2013;63(1):11–30.
Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913–21.
Oh CM, Won YJ, Jung KW, Kong HJ, Cho H, Lee JK, Lee DH, Lee KH, Community of Population-Based Regional Cancer R. Cancer statistics in Korea: incidence, mortality, survival, and prevalence in 2013. Cancer Res Treat. 2016;48(2):436–50.
Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501(7467):328–37.
Namkung J, Kwon W, Choi Y, Yi SG, Han S, Kang MJ, Kim SW, Park T, Jang JY. Molecular subtypes of pancreatic cancer based on miRNA expression profiles have independent prognostic value. J Gastroenterol Hepatol. 2016;31(6):1160–7.
Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. elife. 2015;4:e05005.
Tibshirani R. Regression shrinkage and selection via the Lasso. J Roy Stat Soc B Met. 1996;58(1):267–88.
Zou H, Hastie T. Regularization and variable selection via the elastic net. J R Stat Soc B. 2005;67:301–20.
Meier L, van de Geer SA, Buhlmann P. The group lasso for logistic regression. J R Stat Soc B. 2008;70:53–71.
Kwon MS, Kim Y, Lee S, Namkung J, Yun T, Yi SG, Han S, Kang M, Kim SW, Jang JY, et al. Integrative analysis of multi-omics data for identifying multi-markers for diagnosing pancreatic cancer. BMC Genomics. 2015;16(Suppl 9):S4.
Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125(1–2):279–84.
Yang D, Ma M, Zhou W, Yang B, Xiao C. Inhibition of miR-32 activity promoted EMT induced by PM2.5 exposure through the modulation of the Smad1-mediated signaling pathways in lung cancer cells. Chemosphere. 2017;184:289–98.
Chen R, Liao JY, Huang J, Chen WL, Ma XJ, Luo XD. Downregulation of SRC kinase signaling inhibitor 1 (SRCIN1) expression by MicroRNA-32 promotes proliferation and epithelial-mesenchymal transition in human liver cancer cells. Oncol Res; 2017. https://doi.org/10.3727/096504017X14954923820137.
Chen S, Chen X, Sun KX, Xiu YL, Liu BL, Feng MX, Sang XB, Zhao Y. MicroRNA-93 promotes epithelial-mesenchymal transition of endometrial carcinoma cells. PLoS One. 2016;11(11):e0165776.
Lahdaoui F, Delpu Y, Vincent A, Renaud F, Messager M, Duchene B, Leteurtre E, Mariette C, Torrisani J, Jonckheere N, et al. miR-219-1-3p is a negative regulator of the mucin MUC4 expression and is a tumor suppressor in pancreatic cancer. Oncogene. 2015;34(6):780–8.
Xu L, Li Q, Xu D, Wang Q, An Y, Du Q, Zhang J, Zhu Y, Miao Y. hsa-miR-141 downregulates TM4SF1 to inhibit pancreatic cancer cell invasion and migration. Int J Oncol. 2014;44(2):459–66.
Greither T, Grochola LF, Udelnow A, Lautenschlager C, Wurl P, Taubert H. Elevated expression of microRNAs 155, 203, 210 and 222 in pancreatic tumors is associated with poorer survival. Int J Cancer. 2010;126(1):73–80.
Lee CL, He H, Jiang YJ, Di Y, Yang F, Li J, Jin C, Fu DL. Elevated expression of tumor miR-222 in pancreatic cancer is associated with Ki67 and poor prognosis. Med Oncol. 2013;30(4):700.
Park JK, Henry JC, Jiang J, Esau C, Gusev Y, Lerner MR, Postier RG, Brackett DJ, Schmittgen TD. miR-132 and miR-212 are increased in pancreatic cancer and target the retinoblastoma tumor suppressor. Biochem Biophys Res Commun. 2011;406(4):518–23.
Zhang S, Hao J, Xie F, Hu X, Liu C, Tong J, Zhou J, Wu J, Shao C. Downregulation of miR-132 by promoter methylation contributes to pancreatic cancer development. Carcinogenesis. 2011;32(8):1183–9.
Feng J, Yu J, Pan X, Li Z, Chen Z, Zhang W, Wang B, Yang L, Xu H, Zhang G, et al. HERG1 functions as an oncogene in pancreatic cancer and is downregulated by miR-96. Oncotarget. 2014;5(14):5832–44.
Keklikoglou I, Hosaka K, Bender C, Bott A, Koerner C, Mitra D, Will R, Woerner A, Muenstermann E, Wilhelm H, et al. MicroRNA-206 functions as a pleiotropic modulator of cell proliferation, invasion and lymphangiogenesis in pancreatic adenocarcinoma by targeting ANXA2 and KRAS genes. Oncogene. 2015;34(37):4867–78.
Hu S, Zheng Q, Wu H, Wang C, Liu T, Zhou W. miR-532 promoted gastric cancer migration and invasion by targeting NKD1. Life Sci. 2017;177:15–9.
Bai L, Wang H, Wang AH, Zhang LY, Bai J. MicroRNA-532 and microRNA-3064 inhibit cell proliferation and invasion by acting as direct regulators of human telomerase reverse transcriptase in ovarian cancer. PLoS One. 2017;12(3):e0173912.
Sheikholeslami A, Nabiuni M, Arefian E. Suppressing the molecular signaling pathways involved in inflammation and cancer in breast cancer cell lines MDA-MB-231 and MCF-7 by miR-590. Tumour Biol. 2017;39(4):1010428317697570.
Yang D, Zhao D, Chen X. MiR-133b inhibits proliferation and invasion of gastric cancer cells by up-regulating FBN1 expression. Cancer Biomark. 2017;19:425.
Li D, Xia L, Chen M, Lin C, Wu H, Zhang Y, Pan S, Li X. miR-133b, a particular member of myomiRs, coming into playing its unique pathological role in human cancer. Oncotarget. 2017;8:50193.
Wu H, Wang Y, Wu C, Yang P, Li H, Li Z. Resveratrol induces cancer cell apoptosis through MiR-326/PKM2-mediated ER stress and mitochondrial fission. J Agric Food Chem. 2016;64(49):9356–67.
Ji S, Zhang B, Kong Y, Ma F, Hua Y. MiR-326 inhibits gastric cancer cell growth through down regulating NOB1. Oncol Res. 2017;25(6):53–86.
Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12(12):861–74.
Funding
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI16C2037010016) and Bio-Synergy Research Project of the Ministry of Science, ICT and Future Planning through the National Research Foundation (grant number: 2013M3A9C4078158). Publication of this article was sponsored by the Bio-Synergy Research Project (grant number: 2013M3A9C4078158).
Availability of data and materials
An implementation of HisCoM-mimi, and normalized intensity microarray data can be downloaded from the website (http://statgen.snu.ac.kr/software/hiscom-mimi).
About this supplement
This article has been published as part of BMC Bioinformatics Volume 19 Supplement 4, 2018: Selected articles from the 16th Asia Pacific Bioinformatics Conference (APBC 2018): bioinformatics. The full contents of the supplement are available online at https://bmcbioinformatics.biomedcentral.com/articles/supplements/volume-19-supplement-4.
Author information
Authors and Affiliations
Contributions
YK performed all analyses and developed the software implementation. YK and TP wrote the manuscript and developed the methodology. SL developed the software implementation. SC helped the analysis. JJ provided clinical interpretation of analysis results. All of the authors have read and approved of the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Between the years 2009 and 2012, 200 pancreatic ductal adenocarcinoma (PDAC) samples were collected by the Department of Hepatobiliary and Pancreas Surgery of Seoul National University Hospital. The study protocol was approved by the Institutional Review Board of Seoul National University Hospital (IRB H-0901-010-267) and written, informed consent was obtained from each patient or legally authorized representative.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kim, Y., Lee, S., Choi, S. et al. Hierarchical structural component modeling of microRNA-mRNA integration analysis. BMC Bioinformatics 19 (Suppl 4), 75 (2018). https://doi.org/10.1186/s12859-018-2070-0
Published:
DOI: https://doi.org/10.1186/s12859-018-2070-0