
Abstract
Porous Zn1 − x Mn x O (x = 0.1, 0.2, 0.44) nanosheets were prepared by a low-cost, large-scale production and simple approach, and the applications of these nanosheets as an anode material for Li-ion batteries (LIBs) were explored. Electrochemical measurements showed that the porous Zn0.8Mn0.2O nanosheets still delivered a stable reversible capacity of 210 mA h g−1 at a current rate of 120 mA g−1 up to 300 cycles. These results suggest that the facile synthetic method of producing porous Zn0.8Mn0.2O nanostructure can realize a better cycle durability with stable reversible capacity.
Similar content being viewed by others

Background
ZnO, as 3d a transition-metal oxide, has been considered as an anode material for Li-ion batteries (LIBs) due to the following characteristics: high specific capacity (978 mA h g−1), abundance, low cost, non-toxic, easily produced, and chemically stable [1–5]. However, it often suffers the loss of capacity upon the cycling due to drastic volume changes during the formation of lithium zinc alloys [6–8]. Therefore, a lot of effort has been devoted to overcome these problems; correspondingly, many methods have been developed, such as (i) preparing ordered nanostructured materials [2, 9]; (ii) incorporating with Ni3ZnC0.7 [10]; (iii) coating with Ni, C, and CoO-C layers [6, 11, 12]; and (iv) doping with Mg [13]. These techniques enhance the conductivity, facilitate the lithiation/delithiation process, or buffer the volume changes. For example, Ping et al. reported Zn1 − x Mg x O (x = 0, 0.18) thin films showed an improved cycling stability compared to that of ZnO thin films. The doped Mg ions may only act as a buffer in a form of MgO to alleviate the stress caused by the volume changes during the formation of lithium–zinc alloys [13].
Mn-doped ZnO has been widely investigated for their optical properties, magnetic properties, and sensing properties [14–18]. However, it has rarely been used as anode materials in LIBs. In this work, we successfully synthesized porous Zn1 − x Mn x O (x = 0.1, 0.2, 0.44) nanosheets in high yield using a facile method. The electrode performance of the samples was electrochemically investigated, and the representative as-synthesized Zn1 − x Mn x O (x = 0.2) exhibited a better cycle durability with stable reversible capacity of 210 mA h g−1 for up to 300 cycles at 120 mA g−1.
Methods
-
1.
ZnO precursor synthesis
The ZnO precursor was prepared by our previous reported method [19]. The ZnCl2·5H2O (10.0 mmol) and C6H8O7·H2O (6.7 mmol) were dissolved in 10-mL water, then solid NaOH (50.0 mmol) was directly added into the mixture solution. Subsequently, 40 mL of distilled water was added and a precursor solution was obtained after 10 min. The white precipitate (flower-like ZnO) was obtained.
-
2.
Synthesis of Zn1 − x Mn x O
In a typical process, the obtained ZnO (2.0 mmol) was immersed into Mn(Ac)2·4H2O (0.2 M, 20 mL) aqueous solution and 40 mL of water was added, stirring for 4 days at room temperature. Then, the resulting powders were obtained and washed with distilled water and ethanol several times, dried at 60 °C. Following, the resulting powders were kept at 350 °C for 3 h under argon atmosphere and the target product Zn1 − x Mn x O (x = 0.2) was obtained (denoted as sample A). In addition, Zn1 − x Mn x O (x = 0.1, 0.44) was also obtained with Mn(Ac)2·4H2O with the amount 8 and 30 mL, respectively (denoted as sample B and sample C).
-
3.
Characterization
The as-prepared particles were characterized by powder X-ray diffraction (XRD) on a Philips X’pert X-ray diffractometer equipped with Cu Kα radiation (λ = 1.5418 Å). The morphologies of the samples were examined on a field-emission scanning electron microscopy (FESEM; JEOL JSM-6700 F) and a transmission electron microscopy (TEM; JEOL-2010). The HRTEM images were taken on an aberration-corrected analytical transmission electron microscopy (ARM200F). N2 adsorption–desorption isotherms were measured on a Micromeritics ASAP-2000 nitrogen adsorption apparatus at 150 K.
The electrodes for electrochemical testing consisted of 70 wt% active materials, 15 wt% conductive material (acetylene black), and 15 wt% binder (polyvinylidene fluoride (PVDF)). Test cells (2016) were assembled in glove box using lithium metal as the anode, Celgard 2600 as the separator, and 1 M LiPF6 in ethylene carbonate and dimethyl carbonate solution (v/v, 1:1). The galvanostatical charge/discharge measurement was carried out by a LAND-CT2001 battery cycler (Wuhan, China) testing system in the voltage range of 0.01–3.0 V (vs. Li/Li+).
Results and Discussion
The XRD pattern of the precursor is shown in Fig. 1a. All of the peaks can be assigned to hexagonal ZnO (JCPDS file No. 89-0511). The chemical composition of the sample A was determined by XRD and XPS. The crystallinity and crystal phase of the sample A were demonstrated by the XRD shown in Fig. 1b. The peaks in the diffraction pattern of Zn1 − x Mn x O at 31.81°, 34.43°, and 36.30° can be indexed to a hexagonal wurtzite structure, which consists of three prominent peaks corresponding to (100), (002), and (101) planes, respectively. Compared with the peak position of ZnO (inset), that of Zn1 − x Mn x O was found to shift towards lower angles with Mn incorporation, probably due to the larger ionic radius of Mn2+ (0.066 nm) relative to that of Zn2+ (0.060 nm) [20, 21]. The XPS survey spectrum confirms that the sample mainly contains Zn, Mn, and O (Fig. 2). The strong peaks at around 641.7 eV (Fig. 2a) are assigned to Mn2p3/2. The values correspond to a binding energy of Mn2+ ion [21]. The peak at 1022.4 eV (Fig. 2c) is assigned to Zn2p3/2 for the Zn2+ state (Fig. 2b) and the peak at 531.4 eV (Fig. 2c) corresponds to the binding energy of O1s. In addition, the ratio of Mn to Zn of 6.38:25.05 is given by the quantification of peaks, indicating that the molar ratio of Mn to Zn is near 1:4. The above results indicate that the prepared sample A is single phase Zn0.8Mn0.2O. The SEM images (Fig. 3a, b) show that the Zn0.8Mn0.2O samples are sheet-like morphology with many holes in the nanosheets. From the TEM images (Fig. 3c–e), the porous nanosheets were clearly observed, which further confirms the formation of Zn0.8Mn0.2O porous nanostructures. We also examined the phase purity of the resulting powder that has not been calcined by the XRD. From the XRD pattern (Additional file 1: Figure S1 in supporting information), it is clearly seen that the diffraction pattern of the resulting powder are different from the precursor ZnO and Zn1 − x Mn x O. This result suggests that the chemical reaction process (Mn-doped ZnO) is not simple zinc substituted by manganese but involves a complex reaction which led to its structure and composition being changed. The structure and composition of the resulting powder is complex and difficult to identify. However, due to the similar XRD pattern of Zn(OH)2 and ZnO, it is reasonable that hydroxide ions might be introduced into the resulting powder during the reaction process, in addition to the possible organic molecules. The morphology of resulting powder (Additional file 1: Figure S2) is nanosheets. Interestingly, after calcination, a number of pores appear on the nanosheets, possibly due to decomposition of the organic molecules and hydroxyl group in the calcination process. The pore size distribution curves have been investigated by using Barrett–Joyner–Halenda (BJH) method. Basing on the report of nitrogen adsorption–desorption shown in Additional file 1: Figure S3, the Zn0.8Mn0.2O exhibited a BET surface area of 41.45 m2 g−1 and adsorption average pore diameter of 8.5 nm. For the Zn0.8Mn0.2O nanosheets, lattice images showed fringes with a spacing of ca. 0.1625 nm and ca. 0.2605 nm, corresponding to the (110) and (002) planes of ZnO (Fig. 3f). An overall schematic model of the synthesis procedure is shown in Fig. 4. The energy dispersion X-ray spectrum (EDS) of the as-prepared sample B and C (Additional file 1: Figure S4) shows that the molar ratio of Mn to Zn of 7.99:66.12 and 16.66:20.96 is near 1:8 (Zn0.9Mn0.1O) and 1:1.25 (Zn0.56Mn0.44O), respectively.

a The XRD pattern of the ZnO; b the XRD pattern of the sample A and inset is an enlarged picture of XRD peaks appearing at ~30°–40°

XPS spectra of the sample A: a Mn2p3/2, b Zn2p3/2, and c O1s

SEM images of the sample A: a low magnification, b high magnification; TEM images: c low magnification and d, e high magnification; (f) a HRTEM image

Schematic illustration of the synthesis process of the Zn1 − x Mn x O nanosheets
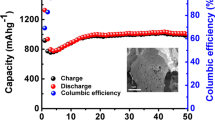
The electrochemical properties of the porous Zn0.8Mn0.2O nanosheets (sample A) were characterized by a galvanostatic method. Figure 5a, b shows first charge/discharge profiles and cycling performance of the Zn0.8Mn0.2O electrode tested in the potential range of 0.01 to 3.0 V and at a current density of 120 mA h g−1. The Zn0.8Mn0.2O nanosheets exhibit the initial discharge and charge capacities of 1198 and 763 mA h g−1, with an initial coulombic efficiency of 64 %. After 50 cycles, this Zn0.8Mn0.2O electrode delivers a reversible capacity of 340 mA h g−1. Even at 300 cycles, this Zn0.8Mn0.2O material still retains a stable capacity of 210 mA h g−1, thus exhibiting an excellent cycle durability. For comparison purposes, we also investigate the electrochemical properties of sample B, sample C, and the ZnO precursor (Fig. 5b). Sample B retains a reversible capacity of ~220 mA h g−1 up to 50 cycle, and the sample delivers a lower capacity of ~90 mA h g−1, while the capacity of the ZnO deteriorates severely and ZnO delivers a much lower capacity of 64 mA h g−1 up to 50 cycles. Apparently, the reversible capacity of the Zn0.8Mn0.2O porous nanosheets is enhanced than that of sample B, sample C, and the precursor ZnO. The good cycle durability of the Zn0.8Mn0.2O material might be reasonable to be attributed to the fact that a great number of the cavities among nanosheets are all beneficial to relieve the strain induced by the severe volume variations of Zn0.8Mn0.2O during Li+ uptake–release, which might improve better rate capacity and the cycling performance.

a Initial discharge/charge profile for the Zn0.8Mn0.2O; b cycling performances and Coulombic efficiencies of the Zn1 − x Mn x O and ZnO electrodes
Conclusions
In conclusion, the Mn-doped ZnO porous nanosheets were successfully synthesized by a simple approach and their electrochemical performance were evaluated. The obtained Zn0.8Mn0.2O porous nanosheets exhibit better cycle durability with good reversible capacity. The cavities among nanosheets maybe could effectively suppress the volume expansion during cycling and enhances the electric conductivity of electrodes, etc., giving rise to better electrochemical performance and cycling stability. In addition, our results provide a simple, effective strategy to fabricate the Zn0.8Mn0.2O nanostructure.
References
Xie Q, Ma Y, Zhang X, Wang L, Yue G, Peng DL. ZnO/Ni/C composite hollow microspheres as anode materials for lithium ion batteries. J Alloys Compd. 2015;619:235–9.
Xiao L, Mei D, Cao M, Qu D, Deng B. Effects of structural patterns and degree of crystallinity on the performance of nanostructured ZnO as anode material for lithium-ion batteries. J Alloys Compd. 2015;627:455–62.
Zhu J, Zhang G, Gu S, Lu B. SnO2 nanorods on ZnO nanofibers: a new class of hierarchical nanostructures enabled by electrospinning as anode material for high-performance lithium-ion batteries. Electrochim Acta. 2014;150:308–13.
Yue HY, Shi ZP, Wang QX, Cao ZX, Dong HY, Qiao Y, et al. MOF-derived cobalt-doped ZnO@C composites as a high-performance anode material for lithium-ion batteries. Acs Appl Mater Inter. 2014;6:17067–74.
Wu MS, Chang HW. Self-assembly of NiO-coated ZnO nanorod electrodes with core–shell nanostructures as anode materials for rechargeable lithium-ion batteries. J Phys Chem C. 2013;117:2590–9.
Zhang CQ, Tu JP, Yuan YF, Huang XH, Chen XT, Mao F. Electrochemical performances of Ni-coated ZnO as an anode material for lithium-ion batteries. J Electrochem Soc. 2007;154:A65–9.
Belliard F, Irvine JTS. Electrochemical performance of ball-milled ZnO-SnO2 systems as anodes in lithium-ion battery. J Power Sources. 2001;97–98:219–22.
Zheng F, Feng H, Ye Z, Yue C, Zong QQ. The electrochemical reaction of zinc oxide thin films with lithium. J Electrochem Soc. 2003;150:A714–20.
Liu J, Li Y, Huang X, Li G, Li Z. Layered double hydroxide nano- and microstructures grown directly on metal substrates and their calcined products for application as Li-ion battery electrodes. Adv Funct Mater. 2008;18:1448–58.
Tang XD, Pan QM, Liu J. Enhancing lithium storage capacity of ZnO anodes through Ni3ZnC0.7 incorporation. J Electrochem Soc. 2010;157:A55–9.
Liu J, Li Y, Ding R, Jiang J, Hu Y, Ji X, et al. Carbon/ZnO nanorod array electrode with significantly improved lithium storage capability. J Phys Chem C. 2009;113:5336–9.
Zhao W, Li QM, Qin PM. Fabrication and electrochemical behavior of flower-like ZnO-CoO-C nanowall arrays as anodes for lithium-ion batteries. J Alloys Compd. 2011;509:9207–13.
Yan GF, Fang HS, Li GS, Li LP, Zhao HJ, Yang Y. Improved electrochemical performance of Mg-doped ZnO thin film as anode material for lithium ion batteries. Chin J Struct Chem. 2009;28:409–13.
Ruan HB, Fang L, Li DC, Saleem M, Qin GP, Kong CY. Effect of dopant concentration on the structural, electrical and optical properties of Mn-doped ZnO films. Thin Solid Films. 2011;519:5078–81.
Xin M, Hu LZ, Liu DP, Yu NS. Effect of Mn doping on the optical, structural and photoluminescence properties of nanostructured ZnO thin film synthesized by sol-gel technique. Superlattice Microst. 2014;74:234–41.
Du SP, Zhao CW, Luo BC, Xing H, Jin KX, Chen CL. Effect of Li-doping on the structure and electrical and magnetic properties of Mn-ZnO film. Rare Metal Mat Eng. 2013;42:1306–9.
Kim JH, Lee JB, Kim H, Choo WK, Ihm Y, Kim D. Electrical and magnetic properties of Mn-doped ZnO. Ferroelectrics. 2002;273:2449–54.
Mao Y, Ma S, Li X, Wang C, Li F, Yang X, et al. Effect of Mn doping on the microstructures and sensing properties of ZnO nanofibers. Appl Sur Sci. 2014;298:109–15.
Wang LL, Zhao DL, Zhang M, Wang CH, Tang KB, Zhang XZ, et al. Zn0.5Co0.5O solid solution nanoparticles with durable life for rechargeable lithium-ion batteries. Nano LIFE. 2014;4:1441015 (7 pages).
Milenova K, Avramova I, Eliyas A, Blaskov V, Stambolova I, Kassabova N. Application of activated M/ZnO (M = Mn, Co, Ni, Cu, Ag) in photocatalytic degradation of diazo textile coloring dye. Environ Sci Pollut R. 2014;21:12249–56.
Ahmad M, Ahmed E, Ahmed W, Elhissi A, Hong ZL, Khalid NR. Enhancing visible light responsive photocatalytic activity by decorating Mn-doped ZnO nanoparticles on grapheme. Ceram Int. 2014;40:10085–97.
Acknowledgements
This work was supported by the Yang Fan project of Science and Technology Commission of Shanghai Municipality (No.14YF1409700) and the Young Teacher Training Scheme of Shanghai (No. ZZgcd14008) and the National Natural Science Foundation of China (No. 21171158, No. 50903018, No 21305086).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing Interests
The authors declare that they have no competing interests.
Authors’ Contributions
LLW prepared the manuscript and carried out the experiment. KBT helped in the technical support for the characterizations and design of the experiment. MZ and JLX participated in the experiment. All the authors discussed the results and approved the final manuscript.
Additional File
Additional file 1:
The XRD pattern of three samples, SEM image of the resulting power, Nitrogen adsorption/desorption isotherms and pore size distribution of Zn 0.8 Mn 0.2 O and EDS spectrum of the products. Figure S1. XRD pattern of three samples, Figure S2. SEM image of the resulting power, Figure S3. Nitrogen adsorption/desorption isotherms and pore size distribution (inset) of Zn0.8Mn0.2O (sample A). Figure S4. EDS spectrum of the products (a) sample B; (b) sample C.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, L., Tang, K., Zhang, M. et al. Facile Synthesis of Mn-Doped ZnO Porous Nanosheets as Anode Materials for Lithium Ion Batteries with a Better Cycle Durability. Nanoscale Res Lett 10, 280 (2015). https://doi.org/10.1186/s11671-015-0983-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11671-015-0983-3