
Abstract
This article reviews current knowledge of the mechanisms underlying the initial hemorrhage and secondary blood–brain barrier (BBB) dysfunction in primary spontaneous intracerebral hemorrhage (ICH) in adults. Multiple etiologies are associated with ICH, for example, hypertension, Alzheimer’s disease, vascular malformations and coagulopathies (genetic or drug-induced). After the initial bleed, there can be continued bleeding over the first 24 hours, so-called hematoma expansion, which is associated with adverse outcomes. A number of clinical trials are focused on trying to limit such expansion. Significant progress has been made on the causes of BBB dysfunction after ICH at the molecular and cell signaling level. Blood components (e.g. thrombin, hemoglobin, iron) and the inflammatory response to those components play a large role in ICH-induced BBB dysfunction. There are current clinical trials of minimally invasive hematoma removal and iron chelation which may limit such dysfunction. Understanding the mechanisms underlying the initial hemorrhage and secondary BBB dysfunction in ICH is vital for developing methods to prevent and treat this devastating form of stroke.
Similar content being viewed by others

Introduction
Each year, there are approximately 800,000 strokes in the USA alone. Although ischemic stroke is the predominant form, approximately 10-15% of these strokes are intracerebral hemorrhage (ICH) and ICH accounts for an even greater percentage of strokes in Asians [1]. ICH is associated with a higher morbidity and mortality compared to ischemic stroke [1]. The occurrence of ICH is also a major concern in the prevention and treatment of ischemia. Thus, anticoagulant treatment for prevention of ischemia (brain and heart) is a growing cause of ICH [2] and the occurrence of symptomatic ICH is a major concern in using tissue plasminogen activator for treating ischemic stroke [3].
Although there are a number of ongoing clinical trials [4], there is currently no FDA approved treatment for ICH. Thus, understanding the mechanisms of brain injury after ICH is important. Vascular disruption is the initial cause of ICH and blood–brain barrier (BBB) dysfunction (with associated edema and leukocyte extravasation) is a secondary consequence [5]. It is vital to understand the mechanisms underlying these two events in ICH.
The aim of this article is to review current knowledge about the mechanisms that may underlie the initial hemorrhage and the secondary BBB dysfunction in adult primary spontaneous ICH. With regards to the latter, there is evidence that cerebral ischemia is not an important component of brain injury after ICH in animal models and in man [5]. Thus, while there is some overlap, there are distinct differences in the mechanisms underlying BBB dysfunction between ischemic stroke and ICH.
During this review, the term BBB dysfunction is used to describe changes in BBB function post-ICH because some of those changes can have deleterious effects (e.g. brain edema may result in brain herniation and death). However, it should be noted that the changes in function may also be adaptive (e.g. infiltrating macrophages may be involved in hematoma resolution). Whether the BBB effects are detrimental or beneficial may depend on hematoma size [6], being beneficial for microbleeds but having adverse effects with large bleeds.
ICH Etiology and hematoma expansion
Hypertension is the most common cause of spontaneous ICH, accounting for about two thirds of hemorrhages. ICH is also linked to amyloid angiopathy, brain tumors and various vascular malformations (e.g. aneurysms, ateriovenous malformations, cerebral cavernous malformations) [7] (Table 1). Increasingly, anticoagulants are also a cause. Their use now accounts for almost 20% of ICH cases in the USA [2]. Location has important impact on the frequency and outcome of ICH. Most ICH is ganglionic or lobar [7] and hindbrain hemorrhages have a worse outcome.
While most bleeding occurs at ictus, in about 30% of ICH patients there is further bleeding (hematoma expansion) during the course of the first day as shown by brain imaging [7, 8]. Such hematoma expansion is associated with poor prognosis and it may, therefore, represent a therapeutic target [8].
Secondary BBB dysfunction
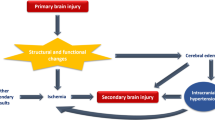
Brain injury and death may result from the initial hemorrhage because of increased intracranial pressure, brain herniation and physical disruption to the brain. However, as in ischemic stroke, ICH also induces secondary brain damage [5]. This includes BBB dysfunction as shown by evidence in animals and humans.
In animals, ICH has mostly been modeled by injecting blood directly into the brain or by intracerebral injection of collagenase. The latter causes bleeding (vascular disruption) via basement membrane degradation. After intracerebral injection of blood into brain, there is delayed BBB hyperpermeability (Figure 1). In rat, Yang et al. found no disruption at 4 hours but progressive disruption from 12 to 48 hours [9]. In pig, Wagner et al. found no disruption from 1 to 8 hours, but found disruption by 24 hours [10, 11]. Following collagenase injection, there was vascular disruption (bleeding) by 30 minutes, but a prolonged lower level BBB hyperpermeability from 5 hours to 7 days [12]. In these animal models, hyperpermeability is associated with brain edema formation and an influx of inflammatory cells into the brain.

Photomicrographs showing both sides of serial coronal sections (thickness approximately 5 mm) containing hematoma (#) and edematous white matter (*) 24 hours after blood infusion in a pig brain frozen in situ. Perihematomal edema is present as blue-staining translucent regions in white matter adjacent to the hematoma. Evans blue staining is observed throughout the ipsilateral white matter and is indicative of increased BBB permeability and vasogenic edema development. Figure/legend reprinted with permission from Journal of Neurosurgery[11].
In human ICH, both acute and delayed vascular disruption occurs. As noted above, in some patients there is continued bleeding and the extravasation of contrast agent during the first 24 hours after ictus [8, 13], but there is also more delayed disruption [14, 15] which may be similar to that found after blood injection in animal models. As in animal models, human BBB disruption after ICH is associated with edema formation (Figure 2) and an influx of leukocytes into brain [5, 7].

Flair magnetic resonance imaging in a patient at day 1 after ICH. Note the marked edema (#) surrounding the hematoma (*).
It is difficult to assess the importance of secondary BBB disruption in ICH. It will contribute to brain edema formation (vasogenic edema), it may participate in neuroinflammation by facilitating leukocyte migration into brain, and it will allow the entry of potentially harmful molecules into brain (e.g. prothrombin which is converted to thrombin and may participate in brain injury; see below). However, while there are experimental treatments that reduce BBB disruption and ameliorate ICH-induced brain injury, those treatments are not ‘BBB-specific’ and it may be that the beneficial effects come from parenchymal cells.
Mechanisms underlying initial hemorrhage
Hypertension
In hypertension, the cause of ICH is thought to relate to remodeling of the cerebral vasculature. Thus, hypertension can cause microaneurysms at the bifurcation of arterioles and chronic elevation of intraluminal arterial pressure can cause small vessel wall damage [16, 17]. Interestingly, in an animal model of ICH, salt fed stroke-prone spontaneously hypertensive rats, BBB hyperpermeability precedes hemorrhaging [18] suggesting there is a gradual weakening of vascular integrity that eventually leads to rupture. There is a genetic component to ICH [19]. Jeanne et al. [20] identified rare mutations in collagen type IV, a component of cerebrovascular basement membranes, that were associated with ICH. Recently, Woo et al. [21], identified 1q22 as a susceptibility locus for non-lobar ICH, although the precise gene involved is still uncertain. Lobar ICH is often due to cerebral amyloid angiopathy and that is discussed below.
Aneurysms
Cerebral aneurysms are balloon-like structures caused by a weakness in a blood vessel wall. It has been estimated that they are present in 3.6-6% of the population over 30 years old [22]. They are prone to rupture. Because of location, bleeding is usually into the subarachnoid space (causing subarachnoid hemorrhage; SAH), but ICH can occur [23]. The risk of bleeding is low in previously unruptured aneurysms, but aneurysm rupture accounts for about 27,000 SAH cases per year in the USA [23].
Cavernous malformations
Cerebral cavernous malformations (CCMs) are mulberry-shaped vascular lesions that occur in the capillary-venous vascular bed [24]. They form vascular channels with little or no intervening parenchymal tissue (Figure 3). The growth (size and number) and bleeding of these lesions can result in neurological symptoms. CCMs are generally sporadic but some are familial (~20%) and, in the latter, loss-of-function mutations have been identified in three genes: CCM1 (KRIT1), CCM2 (malcavernin, OSM, MGC4607) and CCM3 (PDCD10) [24, 25]. CCM1, 2 and 3 can form a complex (the CCM complex signaling platform [25]) and regulate adhesion junction stability [24–26]. The loss of this regulation through mutations may be involved in both lesion growth and bleeding. It should be noted, however, that there are differences in the cellular distribution of CCM1, 2 and 3 [24, 25] and they have important actions outside the CCM complex [24, 25]. Also, there is evidence that CCMs regulate endothelial tight junction function either directly or indirectly via altered adhesion junction function [24]. While there is still much to be learnt about the function of CCM proteins, they do have important effects on endothelial junction proteins, and the loss of those effects that may lead to bleeding.

Hematoxylin & eosin staining of a section from the brain of a patient with cerebral cavernous malformation (type CCM3). Note the multiple blood vessels with little or no intervening parenchymal tissue. Scale bar = 200 μm.
Ateriovenous malformations
Ateriovenous malformations (AVMs) are a tangle of blood vessels lacking a true capillary bed. These are prone to rupture and cerebral hemorrhage is the most severe AVM complication. A number of studies have examined genetic risk factors for hemorrhage. Polymorphisms in the EPHB4 gene encoding a tyrosine kinase receptor involved in arterial-venous determination are associated with bleeding in AVMs [27]. In addition, polymorphisms in the IL-6, TNFα , APOE (the APOEϵ2 allele) and IL-1β genes or gene promoters are associated with hemorrhage in AVMs [28]. As described below, inflammation impacts cerebrovascular integrity.
Amyloid angiopathy
Cerebral amyloid angiopathy (CAA) is the deposition of β-amyloid in small arteries, arterioles and capillaries of the cerebral cortex and leptomeninges [29–31]. β-amyloid is thought to have damaging effects on the vasculature resulting in microbleeds and symptomatic ICH, particularly lobar ICH [31, 32]. CAA occurs in almost all Alzheimer’s disease patients, with severe CAA occurring in about 25% of patients [31]. Patients with the APOEϵ2 or ϵ4 alleles are at greater risk of ICH [31]. Polymorphism in the CR1 gene, encoding the complement component (3b/4b) receptor 1, is associated with development of Alzheimer’s disease, likely because CR1 is involved in β-amyloid clearance [33]. Recently, Biffi et al. found that a variant in the CR1 gene was associated with increased risk of CAA-related ICH [34].
Infection
A recent study raises the interesting possibility that certain types of infection may also cause ICH. Nakano et al. [35] found that infection with a strain of Streptococcus mutans (serotype k) expressing a collagen binding protein (CBP) aggravated cerebral hemorrhage in mice and that the incidence of S. mutans expressing the CBP was higher in hemorrhagic stroke patients than control subjects. Nakano et al. found that CBP-expressing S. mutans accumulated in damaged vessels and that they inhibit collagen-induced platelet activation. The impact of infections with this strain of S. mutans in patients who are susceptible to ICH (e.g. people with CCMs and AVMs) deserves investigation.
Anticoagulants
A growing proportion of ICH cases are linked to anticoagulant use, particularly the vitamin K antagonist warfarin [1, 2]. It has been estimated that there are about 2 million asymptomatic microhemorrhages a year in the USA [36] and the presence of an anticoagulant may transform a microbleed into a symptomatic ICH. The presence of microbleeds increases the occurrence of warfarin-associated ICH by more than 80-fold [37]. Recently, a number of new orally active anticoagulants have become available: dabigatran, a direct thrombin inhibitor, and rivaroxaban and apixaban, Factor Xa antagonists. Initial clinical trials suggest that these may induce less ICH than warfarin [38].
Hemorrhagic transformation
It should be noted that some patients with ischemic stroke will undergo hemorrhagic transformation, i.e. hemorrhaging within the ischemic area [3, 39]. Such hemorrhagic transformation is a particular concern with tissue plasminogen activator (tPA)- induced reperfusion therapy for ischemic stroke. It led to a number of failed clinical trials of thrombolytics, with the adverse effect of increased hemorrhage offsetting the benefit of reperfusion, before the pivotal NINDS clinical trial [40] demonstrated a benefit for tPA. Such hemorrhagic transformation is not the focus of this review on primary spontaneous ICH. The reader is referred to several excellent reviews on vascular dysfunction in hemorrhagic transformation [3, 39, 41].
Mechanisms underlying hematoma expansion
That some patients continue to bleed (hematoma expansion) after the initial ictus, suggests insufficient coagulation. There have, therefore, been attempts to prevent hematoma expansion by manipulating the coagulation cascade. Notably, there was the trial of factor VIIa [42]. That trial found a reduction in hematoma expansion but there was no improvement in outcome. This may have been related to adverse side effects (particularly systemic) of factor VIIa and the fact that only the 30% of patients that undergo hematoma expansion would be expected to benefit. There has, therefore, been interest in identifying which patients are liable to undergo hematoma expansion (e.g. those with the ‘spot sign’ on computerized tomography angiography; [13]) and show most benefit from treatment.
Other techniques that might limit hematoma expansion have been examined. Recently, there has been considerable interest on whether reducing blood pressure will limit hematoma expansion (the INTERACT, ATACH and ICH ADAPT clinical trials, [4, 5]). The results of the phase 3 INTERACT2 trial were reported last year [43]. It showed the apparent safety of acutely lowering blood pressure in ICH patients and suggested a potential benefit on clinical outcome, although this did not quite reach statistical significance. Although the underlying rationale for this (and other) blood pressure lowering trials was to lower hematoma expansion, this phase 3 trial, in contrast to the prior phase 2 trial, failed to show a significant effect of blood pressure reduction on such expansion [43, 44]. Even earlier lowering of blood pressure may have a more beneficial effect.
Mechanisms underlying secondary BBB dysfunction in ICH
The BBB is formed by the cerebral endothelial cells and their linking tight junctions. The tightness of those junctions and a relatively low level of transcytosis results in the BBB having a very low permeability for many compounds unless they are lipophilic or are subject to carrier-mediated transport. In addition, under normal conditions, there is a very low rate of leukocyte migration into brain across the cerebral endothelium. Perivascular cells (e.g. pericytes and astrocytes) and endothelial-extracellular matrix interactions also have a role in regulating BBB function, forming the so called neurovascular unit [45–49]. In addition, leukocytes can also modulate BBB function upon adhesion and during and after transmigration across the endothelium [50].
An increase in BBB permeability and leukocyte infiltration into brain occurs in many neurological conditions. Such an increase may occur via changes in the paracellular (alterations in tight junction function) and/or the transcellular route (transcytosis) across the cerebral endothelium [51]. However, while changes in the para- and transcellular pathways across the endothelium are the ultimate cause of increased permeability after ICH, the effects of ICH may occur either directly on the endothelium or indirectly via effects on other cell types or extracellular matrix that regulate or affect the endothelium. The purpose of this section is to outline what ICH-factors may be involved in inducing BBB dysfunction (hyperpermeability and leukocyte transmigration), the mechanisms involved and the potential impact on ICH-induced brain injury.
ICH induces delayed BBB hyperpermeability [9–11]. A potential cause could be ICH-induced ischemia, with the hematoma mass increasing intracranial pressure and reducing cerebral blood flow (a mass effect), as cerebral ischemia also increases BBB permeability [48]. However, the role of ischemia in ICH-induced brain injury is controversial [5, 7]. Measurements in animal ICH models and a number of human studies indicate that perihematomal blood flows are not markedly reduced and do not reach the levels required to induce ischemic brain damage [52–54]. Indeed, reductions in blood flow may be a result rather than the cause of brain damage (reduced tissue oxygen demand and brain edema may reduce blood flow; [52, 54]). The apparent absence of ICH-induced ischemia suggests that there are either components in blood that increase permeability directly or that the response to those components (e.g. inflammation) causes the hyperpermeability, and that the initiating causes of BBB dysfunction differ from cerebral ischemia (Figure 4). It should be noted, though, that there may be similarities in some of the downstream pathways activated to enhance BBB permeability (e.g. inflammation and tight junction rearrangement).

The underlying cause of secondary BBB dysfunction appears to differ in ICH (A) from cerebral ischemia (B). In ICH, evidence indicates a major role for the presence of blood components in brain parenchyma activating a number of pathways (cell injury, receptor-mediated signaling and inflammation) leading to BBB dysfunction. In contrast, in cerebral ischemia, the initiating cause of injury is the lack of oxygen and glucose supply to the brain. Delayed restoration of blood flow can also induce BBB dysfunction (reperfusion injury). With ischemic injury, there are a number of factors that can enhance the BBB dysfunction. Thus, for example, hyperglycemia and tissue plasminogen activator (tPA) both can result in hemorrhage after reperfusion. Less is known about factors that enhance secondary BBB dysfunction in ICH.
Blood components and secondary blood–brain barrier dysfunction in ICH
Thrombin
Thrombin is generated from plasma prothrombin after an ICH for hemostasis. Intracerebral injection of thrombin causes BBB disruption [55, 56]. Many actions of thrombin are mediated by protease activated receptors (PARs) and particularly PAR-1 [57]. Evidence indicates that BBB disruption by thrombin is at least partly PAR mediated [56, 58] and that thrombin phosphorylates Src kinases to induce endothelial disruption [56]. A PAR-1 antagonist blocked the BBB disruption induced by intraventricular hemorrhage [59]. It should be noted, however, that intracerebral thrombin induces an influx of leukocytes into brain [60] and activates microglia [61] and, as discussed below, inflammation may also cause BBB disruption. Thus, thrombin may have both direct and indirect effects on BBB function.
Fibrin
There may be other components in or derived from plasma involved in BBB disruption and brain injury after ICH. For example, after bleeding or BBB disruption, fibrinogen enters the brain and it is cleaved to form fibrin. In multiple sclerosis, there is evidence that extravascular fibrin is important in neuroinflammation and particularly microglia activation [62, 63]. Although the role of fibrin/fibrinogen in ICH-induced BBB disruption and brain injury has not been studied, microglia activation has been implicated in both (see below). The role of fibrin/fibrinogen and other plasma proteins in ICH deserves further attention.
Erythrocyte components
Erythrocytes are another major component of intracerebral hematomas and the role of erythrocytes and erythrocyte components in ICH-induced injury has received much attention [5, 7, 64]. Erythrocytes within the intracerebral hematoma eventually lyse with hemoglobin release and that hemoglobin can undergo degradation leading to iron release [5, 7, 65] and intracerebral injection of lysed erythrocytes, hemoglobin and iron all cause BBB disruption [66–69].
Although there may be other erythrocyte components involved in ICH-induced brain injury [70], these results suggest that the release and degradation of hemoglobin to iron after erythrocyte lysis contributes to ICH-induced BBB hyperpermeability. This hypothesis is supported by data indicating reduced ICH-induced edema with administration of an iron chelator, deferoxamine [71–73], or a heme oxygenase (HO) inhibitor, zinc protoporphyrin [74, 75]. Heme oxygenases breakdown heme into biliverdin, carbon monoxide, and iron [64]. Two isoforms of HO are present in brain, the constitutive HO-2, and the inducible HO-1 (including very marked induction by ICH [76, 77]). Interestingly, the effects of gene knockout (KO) on ICH-induced brain injury differ for the two isoforms, the HO-1 KO being protective while injury is greater in the HO-2 KO, which may reflect the different cellular distribution of these isoforms [78, 79]. It should also be noted, though, that hemoglobin itself, as well as its degradation products may be involved in inducing BBB dysfunction [68]. There is also evidence that unconjugated bilirubin, a metabolite of biliverdin, can also induce brain edema and inflammation which may be linked to BBB effects [80].
It appears, therefore, that multiple factors produced after erythrocyte lysis can affect BBB function, although iron has a prominent role. Iron is generally thought to cause BBB dysfunction by promoting the generation of free radicals [81, 82]. Oxygen and nitrogen free radicals (e.g. superoxide, hydroxyl and peroxynitrite radicals) can cause endothelial cell damage but also activate cell signaling pathways that regulate BBB permeability [83, 84]. Therapeutically, an iron chelator, deferoxamine, is now in phase II clinical trial for ICH [85].
Inflammation and secondary BBB dysfunction in ICH
Blood components also induce an inflammatory response in the perihematomal area. This includes perihematomal leukocyte infiltration and microglia activation, as well as elevations in cytokines and chemokines and the production and activation of matrix metalloproteinases (MMPs) [7, 86–88]. There is evidence that each of these elements of the inflammatory response can alter BBB function from studies in ICH (see below) and other neurological conditions [50]. In addition, changes in the BBB modulate the inflammatory response. For example, the expression of adhesion molecules on the cerebral endothelium is integral in leukocyte transmigration [50, 89] and infiltrating leukocytes are a major source of cytokines and MMPs after ICH [90, 91].
Neutrophils
In rodent models of ICH, neutrophils are the first leukocytes to invade perihematomal brain tissue, with entry as early as 4 hours and a peak at 48–72 hours [88, 92, 93]. Neutrophils are also found around hemorrhage and in CSF after ICH in patients (reviewed in [88]). In animal models, depleting neutrophils reduces ICH-induced brain injury including secondary BBB disruption [90, 94]. Similarly, toll-like receptor 4 is important in neutrophil entry into brain after ICH and toll-like receptor 4 knockout mice have reduced neutrophil extravasation and reduced ICH-induced brain injury [95]. The toll-like receptor 4-knockout mouse also has reduced macrophage infiltration and microglia activation [96, 97]. Neutrophils are very important sources of TNFα [91] and matrix metalloproteinase-9 (MMP-9) [90] after ICH and may, thereby, influence the entry of other leukocytes into brain and BBB disruption.
Monocytes
Monocytes also invade perihematomal brain tissue. Thus, Hammond et al. [98] found marked invasion of blood-derived monocytes at day 3 after ICH in the mouse, with elevated levels still found at day 7. Two populations of monocytes were detected, CCR2+Ly6Chi and CX3CR1+Ly6C−. The former are inflammatory and they peaked in brain before the latter which may have a role in healing [98]. Depletion of inflammatory monocytes reduced early ICH-induced brain injury. Injury was also reduced in Ccr2-knockout mice and chimeric mice with Ccr2−/− hematopoietic cells [98]. While monocytes may have an adverse effect early in injury, they may also have a role in tissue repair and hematoma phagocytosis later after ICH [86, 98, 99] (see below; Figure 5).

During hematoma resolution after ICH, there is phagocytosis of erythrocytes by invading macrophages/microglia. Hematoxylin & eosin stained section from a pig ICH model (described in [10]) at day 7 after blood injection. Note the phagocytes engulfing erythrocytes (*) and the production of hemosiderin, a breakdown product of hemoglobin (yellow staining).
Lymphocytes
The role of lymphocytes in ICH has received relatively little attention. Xue and Del Bigio [93] noted an influx of CD8a-immunoreactive lymphocytes into the brain between two and seven days after ICH. Peeling et al. [100] found that the immunosuppressant FK-506 (tacrolimus) reduced lymphocyte extravasation after ICH. Rolland et al. [101] found a similar effect with fingolimod, a sphingosine 1-phosphate receptor analog. Both FK-506 and fingolimod also improved functional outcome, although it should be noted that the effects of both drugs were not limited to reducing lymphocyte infiltration into brain. Whether lymphocytes play a role in BBB dysfunction after ICH has not been examined.
In cerebral ischemia and other neurological conditions such as multiple sclerosis, there has been in depth investigation of the mechanisms involved in leukocyte transmigration into brain [50, 102, 103]. Transmigration is a multiple step process in which adhesion molecules on both cerebral endothelial cells and leukocytes play a central role [50, 102, 103]. The mechanism by which neutrophils, macrophages and lymphocytes enter the brain after ICH has received much less attention. Several studies have shown an upregulation of intercellular adhesion molecule-1 (ICAM-1) after experimental ICH [92, 101, 104] and Rolland et al. found that fingolimod blocked both ICAM-1 upregulation and lymphocyte transmigration into brain [101]. Loftspring et al. [80] found that unconjugated bilirubin could also induce ICAM-1 upregulation, suggesting a role of this hemoglobin breakdown product in inflammation after ICH. Ma et al. found upregulation of another adhesion molecule, vascular adhesion protein-1 (VAP-1) after experimental ICH [105] and VAP-1 inhibition had an anti-inflammatory effect [105]. It is likely that multiple adhesion molecules are involved in leukocyte infiltration into brain after ICH.
Microglia
Microglia are rapidly activated after ICH. For example, after intracerebral blood injection in the rat, activated microglia were noted by 4 hours and that activation lasted for at least 4 weeks [93]. A number of studies have shown that acute inhibition of microglia reduces ICH-induced brain injury [87, 88] including BBB disruption [91, 106]. It should be noted, however, that in ICH (as in other neurological conditions), microglia can have beneficial as well as detrimental effects [87, 88], effects which are time-dependent [87]. Thus, microglia are involved in hematoma phagocytosis, tissue repair and can have anti- as well as proinflammatory effects [87, 88]. An alternate therapeutic approach is to modify microglial activation to promote the beneficial effects. Peroxisome proliferator activated receptor (PPAR)-gamma agonists promote the M2 (phagocytic) phenotype in microglia rather than the proinflammatory M1 phenotype and PPAR-gamma agonists have shown benefit in animal ICH models [86, 99]. Pioglitazone, a PPAR-gamma agonist, is now in clinical trial for ICH [107].
Cytokines
Cytokines are signaling molecules that play a central role in inflammatory processes. A variety of cytokines are overexpressed in perihematomal tissue after ICH in animals and man [108, 109] and such changes are also found at the protein level. For example, perihematomal TNFα is markedly increased by 2–4 hours in rat ICH [110] and Sun et al. found prolonged increases in perihematomal IL-1β [111]. Some cytokines are known to increase BBB permeability by activating signaling pathways within the endothelium and by indirect effects on other cells of the neurovascular unit [48, 112, 113]. In ICH, King et al. [114] found a TNFα receptor antagonist could reduce ICH-induced BBB hyperpermeability. Other studies have shown that curcumin [115], erythropoietin [116] and a combination of dexamethasone and melatonin [117] decrease cytokine levels and reduce BBB permeability after ICH.
Chemokines regulate chemotaxis, including the migration of leukocytes. Genomic studies have shown that a range of chemokines are overexpressed in perihematomal tissue after in animal and human ICH [108, 109] and this may relate to the leukocyte influx that occurs after hemorrhage. Ma et al. found protein levels of monocyte chemoattractant protein 1 (CCL2) were increased after experimental ICH [105]. Yao and Tsirka [118] investigated the effects of genetic deletion of CCL2 and its receptor, CCR2, on ICH-induced brain injury. They found that CCL2 or CCR2 deletion reduced hematoma size in a mouse collagenase ICH model, reduced early microglial activation/migration but increased microglia numbers later after ICH. The deletions also reduced leukocyte infiltration. Apart from regulating leukocyte migration and microglial activation, CCL2 can also increase BBB permeability by inducing the internalization of transmembrane tight junction proteins [119–122]. These results suggest that chemokines may play an important role in BBB dysfunction after ICH.
Matrix metalloproteinases
MMPs play a central role in neuroinflammatory processes by degrading elements of the extracellular matrix. Intracerebral injection of bacterial collagenase (a form of MMP) causes hemorrhage [123] and a number of MMPs are upregulated in animal ICH models (e.g. MMP-2, −3, −9 and −12 [124]). MMPs may come from different sources. For example, astrocytes can produce MMP-2, microglia can produce MMP-3 and −12, and neurons can produce MMP-3, but infiltrating neutrophils are a major source of MMP-9 [124], indeed the reduced brain and BBB injury found with neutrophil depletion in rat ICH may be related to reduced MMP-9 levels [90]. There is also evidence for MMP upregulation in ICH patients in blood, CSF and perihematomal tissue (reviewed in [125]). MMP-2 and −9 are upregulated in brains of patients with cerebral amyloid angiopathy including in areas far from the acute ICH [126, 127]. There is, therefore, interest in the role of MMPs in inducing ICH and in secondary brain injury, including BBB disruption.
Although MMPs are best known for extracellular matrix degradation, there is evidence of MMP-mediated cleavage of the BBB tight junction proteins, occludin and claudin-5 [128–131]. Thus, MMPs may affect BBB function and hemorrhage by multiple mechanisms, including disruption of the physical support by the endothelial basement membrane, altered endothelial: extracellular matrix signaling, promotion of leukocyte migration and direct effects on tight junction proteins.
The potential role of MMPs in ICH has been examined in patients by examining whether MMP expression correlates with outcome or markers of injury and in animals by examining the effects of MMP inhibition or genetic deletion on ICH. Thus, several studies have shown that MMP-9 expression correlates with hematoma expansion, perihematomal edema and neurological deterioration in ICH patients (reviewed in [125]). Li et al. recently found that increased plasma MMP-3 and MMP-9 levels were associated with worse outcome in ICH patients but not in brain edema [132]. Animal MMP inhibition/genetic deletion studies in ICH have given some conflicting results. Thus, the broad-based MMP inhibitors GM6001 [133, 134] and BB-1101 [135], and genetic deletion of MMP-12 [136] reduced ICH-induced brain injury. In contrast, another MMP inhibitor, BB-94, increased ICH-induced injury [137], as did genetic deletion of MMP-9 [138]. These divergent effects of MMP manipulation may reflect the diverse roles of different MMPs, roles which may vary with time.
Clinical trials
There are currently a number of clinical trials for ICH therapy (Table 2). As blood components may have deleterious effects on the brain (neurons, glia as well as the BBB), and because the presence of the hematoma may cause physical disruption and increase intracranial pressure, there have been many trials of clot evacuation in ICH (see [139] for a meta-analysis). As yet, these trials have not shown a definitive benefit for surgical removal, including the recent STICH2 trial [140]. This might reflect some of the adverse side effects of surgery. There are currently a number of clinical trials using minimally invasive surgery in combination with clot lysis. An example is the current phase III Minimally Invasive Surgery plus rtPA for Intracerebral Hemorrhage Evacuation (MISTIE) trial which uses t-PA to aid in clot removal [141]. This has recently been reported to reduce perihematomal edema which may suggest reduced BBB permeability [142].
A potential concern of such trials is that surgery or surgery plus tPA might cause the release of factors from the hematoma (e.g. hemoglobin and iron) that are detrimental to the brain parenchyma and the brain vasculature. It is possible that the iron chelator, deferoxamine, which is now in phase II clinical trial for ICH [85], might be of benefit as an adjunct therapy for such clot removal approaches.
As noted above, there are a number of clinical trials focused on acutely reducing blood pressure to limit hematoma expansion [4]. Another pharmacological approach to affect hematoma size is to promote clot resolution. Microglia/macrophages are involved in clot phagocytosis and this is stimulated by PPAR-gamma agonists, such as pioglitazone [86, 99]. It should be noted that PPAR-gamma agonists may have other beneficial effects (anti-inflammatory and anti-oxidant) in ICH. There is a current trial (SHRINC) of pioglitazone in ICH [107].
Conclusion
Intracerebral hemorrhage has a variety of etiologies and the mechanisms causing the initial hemorrhage and subsequent BBB disruption may differ with etiology. Therapeutic approaches may also need to differ (e.g. between hypertensive ICH and that following amyloid angiopathy). However, since our last short review on BBB function in ICH [143], there has been progress made in understanding the mechanisms that underlie initial hemorrhage and secondary BBB dysfunction at the molecular, cellular and tissue level. There are a number of promising current clinical trials for ICH [4] that may limit those processes. Hopefully, the first successful treatments for this devastating form of stroke are not too long in the future.
Abbreviations
- AVMs:
-
Ateriovenous malformations
- BBB:
-
Blood–brain barrier
- CAA:
-
Cerebral amyloid angiopathy
- CBP:
-
Collagen binding protein
- CCMs:
-
Cerebral cavernous malformations
- HO:
-
Heme oxygenase
- ICAM-1:
-
Intercellular adhesion molecule-1
- ICH:
-
Intracerebral hemorrhage
- MMP:
-
Matrix metalloproteinase
- PAR:
-
Protease activated receptor
- PPAR:
-
Peroxisome proliferator activated receptor
- SAH:
-
Subarachnoid hemorrhage
- tPA:
-
Tissue plasminogen activator
- VAP-1:
-
Vascular adhesion protein-1.
References
Adeoye O, Broderick JP: Advances in the management of intracerebral hemorrhage. Nat Rev Neurol. 2010, 6 (11): 593-601.
Flaherty ML: Anticoagulant-associated intracerebral hemorrhage. Semin Neurol. 2010, 30 (5): 565-572.
Jickling GC, Liu D, Stamova B, Ander BP, Zhan X, Lu A, Sharp FR: Hemorrhagic transformation after ischemic stroke in animals and humans. J Cereb Blood Flow Metab. 2014, 34 (2): 185-199.
Xi G, Strahle J, Hua Y, Keep RF: Progress in translational research on intracerebral hemorrhage: is there an end in sight?. Prog Neurobiol. 2014, 115: 45-63.
Keep RF, Hua Y, Xi G: Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol. 2012, 11 (8): 720-731.
Keep RF, Xi G, Hua Y, Hoff JT: The deleterious or beneficial effects of different agents in intracerebral hemorrhage: think big, think small, or is hematoma size important?. Stroke. 2005, 36 (7): 1594-1596.
Xi G, Keep RF, Hoff JT: Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006, 5 (1): 53-63.
Brouwers HB, Greenberg SM: Hematoma expansion following acute intracerebral hemorrhage. Cerebrovasc Dis. 2013, 35 (3): 195-201.
Yang GY, Betz AL, Chenevert TL, Brunberg JA, Hoff JT: Experimental intracerebral hemorrhage: relationship between brain edema, blood flow, and blood–brain barrier permeability in rats. J Neurosurg. 1994, 81 (1): 93-102.
Wagner KR, Xi G, Hua Y, Kleinholz M, de Courten-Myers GM, Myers RE, Broderick JP, Brott TG: Lobar intracerebral hemorrhage model in pigs: rapid edema development in perihematomal white matter. Stroke. 1996, 27 (3): 490-497.
Wagner KR, Xi G, Hua Y, Zuccarello M, de Courten-Myers GM, Broderick JP, Brott TG: Ultra-early clot aspiration after lysis with tissue plasminogen activator in a porcine model of intracerebral hemorrhage: edema reduction and blood–brain barrier protection. J Neurosurg. 1999, 90 (3): 491-498.
Rosenberg GA, Estrada E, Kelley RO, Kornfeld M: Bacterial collagenase disrupts extracellular matrix and opens blood–brain barrier in rat. Neurosci Lett. 1993, 160 (1): 117-119.
Hallevi H, Abraham AT, Barreto AD, Grotta JC, Savitz SI: The spot sign in intracerebral hemorrhage: the importance of looking for contrast extravasation. Cerebrovasc Dis. 2010, 29 (3): 217-220.
Delgado P, Alvarez Sabin J, Santamarina E, Molina CA, Quintana M, Rosell A, Montaner J: Plasma S100B level after acute spontaneous intracerebral hemorrhage. Stroke. 2006, 37 (11): 2837-2839.
Murai Y, Ikeda Y, Teramoto A, Tsuji Y: Magnetic resonance imaging-documented extravasation as an indicator of acute hypertensive intracerebral hemorrhage. J Neurosurg. 1998, 88 (4): 650-655.
Fisher CM: Pathological observations in hypertensive cerebral hemorrhage. J Neuropath Exp Neurol. 1971, 30 (3): 536-550.
Fisher CM: Cerebral miliary aneurysms in hypertension. Am J Pathol. 1972, 66 (2): 313-330.
Lee JM, Zhai G, Liu Q, Gonzales ER, Yin K, Yan P, Hsu CY, Vo KD, Lin W: Vascular permeability precedes spontaneous intracerebral hemorrhage in stroke-prone spontaneously hypertensive rats. Stroke. 2007, 38 (12): 3289-3291.
Devan WJ, Falcone GJ, Anderson CD, Jagiella JM, Schmidt H, Hansen BM, Jimenez-Conde J, Giralt-Steinhauer E, Cuadrado-Godia E, Soriano C, Ayres AM, Schwab K, Kassis SB, Valant V, Pera J, Urbanik A, Viswanathan A, Rost NS, Goldstein JN, Freudenberger P, Stogerer EM, Norrving B, Tirschwell DL, Selim M, Brown DL, Silliman SL, Worrall BB, Meschia JF, Kidwell CS, Montaner J: Heritability estimates identify a substantial genetic contribution to risk and outcome of intracerebral hemorrhage. Stroke. 2013, 44 (6): 1578-1583.
Jeanne M, Labelle-Dumais C, Jorgensen J, Kauffman WB, Mancini GM, Favor J, Valant V, Greenberg SM, Rosand J, Gould DB: COL4A2 mutations impair COL4A1 and COL4A2 secretion and cause hemorrhagic stroke. Am J Hum Genet. 2012, 90 (1): 91-101.
Woo D, Falcone GJ, Devan WJ, Brown WM, Biffi A, Howard TD, Anderson CD, Brouwers HB, Valant V, Battey TW, Enzinger C, Hansen BM, Norrving B, Jimenez-Conde J, Giralt-Steinhauer E, Elosua R, Cuadrado-Godia E, Soriano C, Roquer J, Kraft P, Ayres AM, Schwab K, McCauley JL, Pera J, Urbanik A, Rost NS, Goldstein JN, Viswanathan A, Stogerer EM, Tirschwell DL, et al: Meta-analysis of genome-wide association studies identifies 1q22 as a susceptibility locus for intracerebral hemorrhage. Am J Hum Genet. 2014, 94 (4): 511-521.
Penn DL, Komotar RJ, Sander Connolly E: Hemodynamic mechanisms underlying cerebral aneurysm pathogenesis. J Clin Neurosci. 2011, 18 (11): 1435-1438.
McNulty ML, Lee VH: Management of unruptured intracranial aneurysms and arteriovenous malformations. Am J Ther. 2011, 18 (1): 64-69.
Fischer A, Zalvide J, Faurobert E, Albiges-Rizo C, Tournier-Lasserve E: Cerebral cavernous malformations: from CCM genes to endothelial cell homeostasis. Trends Mol Med. 2013, 19 (5): 302-308.
Fisher OS, Boggon TJ: Signaling pathways and the cerebral cavernous malformations proteins: lessons from structural biology. Cell Mol Life Sci. 2014, 71 (10): 1881-1892.
Dejana E, Orsenigo F: Endothelial adherens junctions at a glance. J Cell Sci. 2013, 126 (Pt 12): 2545-2549.
Weinsheimer S, Kim H, Pawlikowska L, Chen Y, Lawton MT, Sidney S, Kwok PY, McCulloch CE, Young WL: EPHB4 gene polymorphisms and risk of intracranial hemorrhage in patients with brain arteriovenous malformations. Circ Cardiovasc Genet. 2009, 2 (5): 476-482.
Rutledge WC, Ko NU, Lawton MT, Kim H: Hemorrhage rates and risk factors in the natural history course of brain arteriovenous malformations. Transl Stroke Res. 2014, 5 (5): 538-542.
Auriel E, Greenberg SM: The pathophysiology and clinical presentation of cerebral amyloid angiopathy. Curr Atheroscler Rep. 2012, 14 (4): 343-350.
Mehndiratta P, Manjila S, Ostergard T, Eisele S, Cohen ML, Sila C, Selman WR: Cerebral amyloid angiopathy-associated intracerebral hemorrhage: pathology and management. Neurosurg Focus. 2012, 32 (4): E7-
Viswanathan A, Greenberg SM: Cerebral amyloid angiopathy in the elderly. Ann Neurol. 2011, 70 (6): 871-880.
Samarasekera N, Smith C, Al-Shahi Salman R: The association between cerebral amyloid angiopathy and intracerebral haemorrhage: systematic review and meta-analysis. J Neurol Neurosurg Psych. 2012, 83 (3): 275-281.
Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fievet N, Barberger-Gateau P, Engelborghs S, De Deyn P, Mateo I, Franck A, Helisalmi S, Porcellini E, Hanon O: Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009, 41 (10): 1094-1099.
Biffi A, Shulman JM, Jagiella JM, Cortellini L, Ayres AM, Schwab K, Brown DL, Silliman SL, Selim M, Worrall BB, Meschia JF, Slowik A, De Jager PL, Greenberg SM, Schneider JA, Bennett DA, Rosand J: Genetic variation at CR1 increases risk of cerebral amyloid angiopathy. Neurology. 2012, 78 (5): 334-341.
Nakano K, Hokamura K, Taniguchi N, Wada K, Kudo C, Nomura R, Kojima A, Naka S, Muranaka Y, Thura M, Nakajima A, Masuda K, Nakagawa I, Speziale P, Shimada N, Amano A, Kamisaki Y, Tanaka T, Umemura K, Ooshima T: The collagen-binding protein of Streptococcus mutans is involved in haemorrhagic stroke. Nat Commun. 2011, 2: 485-
Leary MC, Saver JL: Annual incidence of first silent stroke in the United States: a preliminary estimate. Cerebrovasc Dis. 2003, 16 (3): 280-285.
Lee SH, Ryu WS, Roh JK: Cerebral microbleeds are a risk factor for warfarin-related intracerebral hemorrhage. Neurology. 2009, 72 (2): 171-176.
Lauer A, Pfeilschifter W, Schaffer CB, Lo EH, Foerch C: Intracerebral haemorrhage associated with antithrombotic treatment: translational insights from experimental studies. Lancet Neurol. 2013, 12 (4): 394-405.
Khatri R, McKinney AM, Swenson B, Janardhan V: Blood–brain barrier, reperfusion injury, and hemorrhagic transformation in acute ischemic stroke. Neurology. 2012, 79 (13 Suppl 1): S52-S57.
Anonymous: Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. New Engl J Med. 1995, 333 (24): 1581-1587.
del Zoppo GJ, Izawa Y, Hawkins BT: Hemostasis and alterations of the central nervous system. Semin Thromb Hemost. 2013, 39 (8): 856-875.
Mayer SA, Brun NC, Begtrup K, Broderick J, Davis S, Diringer MN, Skolnick BE, Steiner T, Investigators FT: Efficacy and safety of recombinant activated factor VII for acute intracerebral hemorrhage. New Engl J Med. 2008, 358 (20): 2127-2137.
Anderson CS, Heeley E, Huang Y, Wang J, Stapf C, Delcourt C, Lindley R, Robinson T, Lavados P, Neal B, Hata J, Arima H, Parsons M, Li Y, Wang J, Heritier S, Li Q, Woodward M, Simes RJ, Davis SM, Chalmers J: INTERACT Investigators: Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. New Engl J Med. 2013, 368 (25): 2355-2365.
Anderson CS, Huang Y, Arima H, Heeley E, Skulina C, Parsons MW, Peng B, Li Q, Su S, Tao QL, Li YC, Jiang JD, Tai LW, Zhang JL, Xu E, Cheng Y, Morgenstern LB, Chalmers J, Wang JG: INTERACT Investigators: effects of early intensive blood pressure-lowering treatment on the growth of hematoma and perihematomal edema in acute intracerebral hemorrhage: the Intensive Blood Pressure Reduction in Acute Cerebral Haemorrhage Trial (INTERACT). Stroke. 2010, 41 (2): 307-312.
Abbott NJ, Ronnback L, Hansson E: Astrocyte-endothelial interactions at the blood–brain barrier. Nat Rev Neurosci. 2006, 7 (1): 41-53.
del Zoppo GJ: Aging and the neurovascular unit. Ann New York Acad Sci. 2012, 1268: 127-133.
Mae M, Armulik A, Betsholtz C: Getting to know the cast - cellular interactions and signaling at the neurovascular unit. Curr Pharm Design. 2011, 17 (26): 2750-2754.
Ronaldson PT, Davis TP: Blood–brain barrier integrity and glial support: mechanisms that can be targeted for novel therapeutic approaches in stroke. Curr Pharm Design. 2012, 18 (25): 3624-3644.
Stanimirovic DB, Friedman A: Pathophysiology of the neurovascular unit: disease cause or consequence?. J Cereb Blood Flow Metab. 2012, 32 (7): 1207-1221.
Greenwood J, Heasman SJ, Alvarez JI, Prat A, Lyck R, Engelhardt B: Review: leucocyte-endothelial cell crosstalk at the blood–brain barrier: a prerequisite for successful immune cell entry to the brain. Neuropath Appl Neuro. 2011, 37 (1): 24-39.
Knowland D, Arac A, Sekiguchi KJ, Hsu M, Lutz SE, Perrino J, Steinberg GK, Barres BA, Nimmerjahn A, Agalliu D: Stepwise recruitment of transcellular and paracellular pathways underlies blood–brain barrier breakdown in stroke. Neuron. 2014, 82 (3): 603-617.
Herweh C, Juttler E, Schellinger PD, Klotz E, Schramm P: Perfusion CT in hyperacute cerebral hemorrhage within 3 hours after symptom onset: is there an early perihemorrhagic penumbra?. J Neuroimaging. 2010, 20 (4): 350-353.
Kate MP, Hansen MB, Mouridsen K, Ostergaard L, Choi V, Gould BE, McCourt R, Hill MD, Demchuk AM, Coutts SB, Dowlatshahi D, Emery DJ, Buck BH, Butcher KS: ICHADAPT Investigators: blood pressure reduction does not reduce perihematoma oxygenation: a CT perfusion study. J Cereb Blood Flow Metab. 2014, 34 (1): 81-86.
Zazulia AR, Diringer MN, Videen TO, Adams RE, Yundt K, Aiyagari V, Grubb RL, Powers WJ: Hypoperfusion without ischemia surrounding acute intracerebral hemorrhage. J Cereb Blood Flow Metab. 2001, 21 (7): 804-810.
Lee KR, Kawai N, Kim S, Sagher O, Hoff JT: Mechanisms of edema formation after intracerebral hemorrhage: effects of thrombin on cerebral blood flow, blood–brain barrier permeability, and cell survival in a rat model. J Neurosurg. 1997, 86 (2): 272-278.
Liu DZ, Ander BP, Xu H, Shen Y, Kaur P, Deng W, Sharp FR: Blood–brain barrier breakdown and repair by Src after thrombin-induced injury. Ann Neurol. 2010, 67 (4): 526-533.
Xi G, Reiser G, Keep RF: The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: deleterious or protective?. J Neurochem. 2003, 84 (1): 3-9.
Yan J, Manaenko A, Chen S, Klebe D, Ma Q, Caner B, Fujii M, Zhou C, Zhang JH: Role of SCH79797 in maintaining vascular integrity in rat model of subarachnoid hemorrhage. Stroke. 2013, 44 (5): 1410-1417.
Gao F, Liu F, Chen Z, Hua Y, Keep RF, Xi G: Hydrocephalus after intraventricular hemorrhage: the role of thrombin. J Cereb Blood Flow Metab. 2014, 34 (3): 489-494.
Masada T, Hua Y, Xi G, Yang GY, Hoff JT, Keep RF: Attenuation of intracerebral hemorrhage and thrombin-induced brain edema by overexpression of interleukin-1 receptor antagonist. J Neurosurg. 2001, 95 (4): 680-686.
Moller T, Weinstein JR, Hanisch UK: Activation of microglial cells by thrombin: past, present, and future. Semin Thromb Hemost. 2006, 32 (Suppl 1): 69-76.
Adams RA, Passino M, Sachs BD, Nuriel T, Akassoglou K: Fibrin mechanisms and functions in nervous system pathology. Mol Interv. 2004, 4 (3): 163-176.
Davalos D, Ryu JK, Merlini M, Baeten KM, Le Moan N, Petersen MA, Deerinck TJ, Smirnoff DS, Bedard C, Hakozaki H, Gonias MS, Ling JB, Lassmann H, Degen JL, Ellisman MH, Akassoglou K: Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun. 2012, 3: 1227-
Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF: Heme and iron metabolism: role in cerebral hemorrhage. J Cereb Blood Flow Metab. 2003, 23 (6): 629-652.
Hua Y, Keep RF, Hoff JT, Xi G: Brain injury after intracerebral hemorrhage: the role of thrombin and iron. Stroke. 2007, 38 (2 Suppl): 759-762.
Katsu M, Niizuma K, Yoshioka H, Okami N, Sakata H, Chan PH: Hemoglobin-induced oxidative stress contributes to matrix metalloproteinase activation and blood–brain barrier dysfunction in vivo. J Cereb Blood Flow Metab. 2010, 30 (12): 1939-1950.
Xi G, Hua Y, Bhasin RR, Ennis SR, Keep RF, Hoff JT: Mechanisms of edema formation after intracerebral hemorrhage: effects of extravasated red blood cells on blood flow and blood–brain barrier integrity. Stroke. 2001, 32 (12): 2932-2938.
Yang S, Chen Y, Deng X, Jiang W, Li B, Fu Z, Du M, Ding R: Hemoglobin-induced nitric oxide synthase overexpression and nitric oxide production contribute to blood–brain barrier disruption in the rat. J Mol Neurosci. 2013, 51 (2): 352-363.
Zhao F, Hua Y, He Y, Keep RF, Xi G: Minocycline-induced attenuation of iron overload and brain injury after experimental intracerebral hemorrhage. Stroke. 2011, 42 (12): 3587-3593.
Guo F, Hua Y, Wang J, Keep RF, Xi G: Inhibition of carbonic anhydrase reduces brain injury after intracerebral hemorrhage. Transl Stroke Res. 2012, 3 (1): 130-137.
Nakamura T, Keep RF, Hua Y, Schallert T, Hoff JT, Xi G: Deferoxamine-induced attenuation of brain edema and neurological deficits in a rat model of intracerebral hemorrhage. J Neurosurg. 2004, 100 (4): 672-678.
Okauchi M, Hua Y, Keep RF, Morgenstern LB, Schallert T, Xi G: Deferoxamine treatment for intracerebral hemorrhage in aged rats: therapeutic time window and optimal duration. Stroke. 2010, 41 (2): 375-382.
Xie Q, Gu Y, Hua Y, Liu W, Keep RF, Xi G: Deferoxamine attenuates white matter injury in a piglet intracerebral hemorrhage model. Stroke. 2014, 45 (1): 290-292.
Gong Y, Tian H, Xi G, Keep RF, Hoff JT, Hua Y: Systemic zinc protoporphyrin administration reduces intracerebral hemorrhage-induced brain injury. Acta Neurochir Suppl. 2006, 96: 232-236.
Wagner KR, Hua Y, de Courten-Myers GM, Broderick JP, Nishimura RN, Lu SY, Dwyer BE: Tin-mesoporphyrin, a potent heme oxygenase inhibitor, for treatment of intracerebral hemorrhage: in vivo and in vitro studies. Cell Mol Biol. 2000, 46 (3): 597-608.
Matz PG, Weinstein PR, Sharp FR: Heme oxygenase-1 and heat shock protein 70 induction in glia and neurons throughout rat brain after experimental intracerebral hemorrhage. Neurosurgery. 1997, 40 (1): 152-160.
Wu J, Hua Y, Keep RF, Nakamura T, Hoff JT, Xi G: Iron and iron-handling proteins in the brain after intracerebral hemorrhage. Stroke. 2003, 34 (12): 2964-2969.
Wang J, Dore S: Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage. Brain. 2007, 130 (Pt 6): 1643-1652.
Wang J, Dore S: Heme oxygenase 2 deficiency increases brain swelling and inflammation after intracerebral hemorrhage. Neuroscience. 2008, 155 (4): 1133-1141.
Loftspring MC, Johnson HL, Feng R, Johnson AJ, Clark JF: Unconjugated bilirubin contributes to early inflammation and edema after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2011, 31 (4): 1133-1142.
Chan PH, Schmidley JW, Fishman RA, Longar SM: Brain injury, edema, and vascular permeability changes induced by oxygen-derived free radicals. Neurology. 1984, 34 (3): 315-320.
Zuccarello M, Anderson DK: Interaction between free radicals and excitatory amino acids in the blood–brain barrier disruption after iron injury in the rat. J Neurotraum. 1993, 10 (4): 397-403.
Fraser PA: The role of free radical generation in increasing cerebrovascular permeability. Free Radical Bio Med. 2011, 51 (5): 967-977.
Pun PB, Lu J, Moochhala S: Involvement of ROS in BBB dysfunction. Free Radical Res. 2009, 43 (4): 348-364.
Yeatts SD, Palesch YY, Moy CS, Selim M: High dose deferoxamine in intracerebral hemorrhage (HI-DEF) trial: rationale, design, and methods. Neurocrit Care. 2013, 19 (2): 257-266.
Aronowski J, Zhao X: Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke. 2011, 42 (6): 1781-1786.
Taylor RA, Sansing LH: Microglial responses after ischemic stroke and intracerebral hemorrhage. Clin Devel Immunol. 2013, 2013: 746068-
Wang J, Dore S: Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2007, 27 (5): 894-908.
Engelhardt B: Immune cell entry into the central nervous system: involvement of adhesion molecules and chemokines. J Neurol Sci. 2008, 274 (1–2): 23-26.
Moxon-Emre I, Schlichter LC: Neutrophil depletion reduces blood–brain barrier breakdown, axon injury, and inflammation after intracerebral hemorrhage. J Neuropath Exp Neurol. 2011, 70 (3): 218-235.
Wasserman JK, Schlichter LC: Minocycline protects the blood–brain barrier and reduces edema following intracerebral hemorrhage in the rat. Exp Neurol. 2007, 207 (2): 227-237.
Gong C, Hoff JT, Keep RF: Acute inflammatory reaction following experimental intracerebral hemorrhage in rat. Brain Res. 2000, 871 (1): 57-65.
Xue M, Del Bigio MR: Intracerebral injection of autologous whole blood in rats: time course of inflammation and cell death. Neurosci Lett. 2000, 283 (3): 230-232.
Sansing LH, Harris TH, Kasner SE, Hunter CA, Kariko K: Neutrophil depletion diminishes monocyte infiltration and improves functional outcome after experimental intracerebral hemorrhage. Acta Neurochir Suppl. 2011, 111: 173-178.
Sansing LH, Harris TH, Welsh FA, Kasner SE, Hunter CA, Kariko K: Toll-like receptor 4 contributes to poor outcome after intracerebral hemorrhage. Ann Neurol. 2011, 70 (4): 646-656.
Fang H, Wang PF, Zhou Y, Wang YC, Yang QW: Toll-like receptor 4 signaling in intracerebral hemorrhage-induced inflammation and injury. J Neuroinflamm. 2013, 10: 27-
Lin S, Yin Q, Zhong Q, Lv FL, Zhou Y, Li JQ, Wang JZ, Su BY, Yang QW: Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J Neuroinflamm. 2012, 9: 46-
Hammond MD, Taylor RA, Mullen MT, Ai Y, Aguila HL, Mack M, Kasner SE, McCullough LD, Sansing LH: CCR2+ Ly6C(hi) inflammatory monocyte recruitment exacerbates acute disability following intracerebral hemorrhage. J Neurosci. 2014, 34 (11): 3901-3909.
Zhao X, Grotta J, Gonzales N, Aronowski J: Hematoma resolution as a therapeutic target: the role of microglia/macrophages. Stroke. 2009, 40 (3 Suppl): S92-S94.
Peeling J, Yan HJ, Corbett D, Xue M, Del Bigio MR: Effect of FK-506 on inflammation and behavioral outcome following intracerebral hemorrhage in rat. Exp Neurol. 2001, 167 (2): 341-347.
Rolland WB, Lekic T, Krafft PR, Hasegawa Y, Altay O, Hartman R, Ostrowski R, Manaenko A, Tang J, Zhang JH: Fingolimod reduces cerebral lymphocyte infiltration in experimental models of rodent intracerebral hemorrhage. Exp Neurol. 2013, 241: 45-55.
Engelhardt B, Ransohoff RM: Capture, crawl, cross: the T cell code to breach the blood–brain barriers. Trend Immunol. 2012, 33 (12): 579-589.
Larochelle C, Alvarez JI, Prat A: How do immune cells overcome the blood–brain barrier in multiple sclerosis?. FEBS Lett. 2011, 585 (23): 3770-3780.
Yang JT, Lee TH, Lee IN, Chung CY, Kuo CH, Weng HH: Dexamethasone inhibits ICAM-1 and MMP-9 expression and reduces brain edema in intracerebral hemorrhagic rats. Acta Neurochir. 2011, 153 (11): 2197-2203.
Ma Q, Manaenko A, Khatibi NH, Chen W, Zhang JH, Tang J: Vascular adhesion protein-1 inhibition provides antiinflammatory protection after an intracerebral hemorrhagic stroke in mice. J Cereb Blood Flow Metab. 2011, 31 (3): 881-893.
Shi W, Wang Z, Pu J, Wang R, Guo Z, Liu C, Sun J, Gao L, Zhou R: Changes of blood–brain barrier permeability following intracerebral hemorrhage and the therapeutic effect of minocycline in rats. Acta Neurochir Suppl. 2011, 110 (Pt 2): 61-67.
Gonzales NR, Shah J, Sangha N, Sosa L, Martinez R, Shen L, Kasam M, Morales MM, Hossain MM, Barreto AD, Savitz SI, Lopez G, Misra V, Wu TC, El Khoury R, Sarraj A, Sahota P, Hicks W, Acosta I, Sline MR, Rahbar MH, Zhao X, Aronowski J, Grotta JC: Design of a prospective, dose-escalation study evaluating the Safety of Pioglitazone for Hematoma Resolution in Intracerebral Hemorrhage (SHRINC). Int J Stroke. 2013, 8 (5): 388-396.
Lu A, Tang Y, Ran R, Ardizzone TL, Wagner KR, Sharp FR: Brain genomics of intracerebral hemorrhage. J Cereb Blood Flow Metab. 2006, 26 (2): 230-252.
Rosell A, Vilalta A, Garcia-Berrocoso T, Fernandez-Cadenas I, Domingues-Montanari S, Cuadrado E, Delgado P, Ribo M, Martinez-Saez E, Ortega-Aznar A, Montaner J: Brain perihematoma genomic profile following spontaneous human intracerebral hemorrhage. PLoS One. 2011, 6 (2): e16750-
Hua Y, Wu J, Keep RF, Nakamura T, Hoff JT, Xi G: Tumor necrosis factor-alpha increases in the brain after intracerebral hemorrhage and thrombin stimulation. Neurosurgery. 2006, 58 (3): 542-550.
Sun H, Tang Y, Guan X, Li L, Wang D: Effects of selective hypothermia on blood–brain barrier integrity and tight junction protein expression levels after intracerebral hemorrhage in rats. Biol Chem. 2013, 394 (10): 1317-1324.
Banks WA, Erickson MA: The blood–brain barrier and immune function and dysfunction. Neurobiol Dis. 2010, 37 (1): 26-32.
Pan W, Stone KP, Hsuchou H, Manda VK, Zhang Y, Kastin AJ: Cytokine signaling modulates blood–brain barrier function. Curr Pharm Design. 2011, 17 (33): 3729-3740.
King MD, Alleyne CH, Dhandapani KM: TNF-alpha receptor antagonist, R-7050, improves neurological outcomes following intracerebral hemorrhage in mice. Neurosci Lett. 2013, 542: 92-96.
King MD, McCracken DJ, Wade FM, Meiler SE, Alleyne CH, Dhandapani KM: Attenuation of hematoma size and neurological injury with curcumin following intracerebral hemorrhage in mice. J Neurosurg. 2011, 115 (1): 116-123.
Chau M, Chen D, Wei L: Erythropoietin attenuates inflammatory factors and cell death in neonatal rats with intracerebral hemorrhage. Acta Neurochir Suppl. 2011, 111: 299-305.
Li ZQ, Liang GB, Xue YX, Liu YH: Effects of combination treatment of dexamethasone and melatonin on brain injury in intracerebral hemorrhage model in rats. Brain Res. 2009, 1264: 98-103.
Yao Y, Tsirka SE: The CCL2-CCR2 system affects the progression and clearance of intracerebral hemorrhage. GLIA. 2012, 60 (6): 908-918.
Stamatovic SM, Dimitrijevic OB, Keep RF, Andjelkovic AV: Protein kinase Calpha-RhoA cross-talk in CCL2-induced alterations in brain endothelial permeability. J Biol Chem. 2006, 281 (13): 8379-8388.
Stamatovic SM, Keep RF, Kunkel SL, Andjelkovic AV: Potential role of MCP-1 in endothelial cell tight junction ‘opening’: signaling via Rho and Rho kinase. J Cell Sci. 2003, 116 (Pt 22): 4615-4628.
Stamatovic SM, Keep RF, Wang MM, Jankovic I, Andjelkovic AV: Caveolae-mediated internalization of occludin and claudin-5 during CCL2-induced tight junction remodeling in brain endothelial cells. J Biol Chem. 2009, 284 (28): 19053-19066.
Stamatovic SM, Shakui P, Keep RF, Moore BB, Kunkel SL, Van Rooijen N, Andjelkovic AV: Monocyte chemoattractant protein-1 regulation of blood–brain barrier permeability. J Cereb Blood Flow Metab. 2005, 25 (5): 593-606.
Rosenberg GA, Mun-Bryce S, Wesley M, Kornfeld M: Collagenase-induced intracerebral hemorrhage in rats. Stroke. 1990, 21 (5): 801-807.
Xue M, Yong VW: Matrix metalloproteinases in intracerebral hemorrhage. Neurol Res. 2008, 30 (8): 775-782.
Florczak-Rzepka M, Grond-Ginsbach C, Montaner J, Steiner T: Matrix metalloproteinases in human spontaneous intracerebral hemorrhage: an update. Cerebrovasc Dis. 2012, 34 (4): 249-262.
Hartz AM, Bauer B, Soldner EL, Wolf A, Boy S, Backhaus R, Mihaljevic I, Bogdahn U, Klunemann HH, Schuierer G, Schlachetzki F: Amyloid-beta contributes to blood–brain barrier leakage in transgenic human amyloid precursor protein mice and in humans with cerebral amyloid angiopathy. Stroke. 2012, 43 (2): 514-523.
Hernandez-Guillamon M, Martinez-Saez E, Delgado P, Domingues-Montanari S, Boada C, Penalba A, Boada M, Pagola J, Maisterra O, Rodriguez-Luna D, Molina CA, Rovira A, Alvarez-Sabin J, Ortega-Aznar A, Montaner J: MMP-2/MMP-9 plasma level and brain expression in cerebral amyloid angiopathy-associated hemorrhagic stroke. Brain Pathol. 2012, 22 (2): 133-141.
Lischper M, Beuck S, Thanabalasundaram G, Pieper C, Galla HJ: Metalloproteinase mediated occludin cleavage in the cerebral microcapillary endothelium under pathological conditions. Brain Res. 2010, 1326: 114-127.
Liu J, Jin X, Liu KJ, Liu W: Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood–brain barrier damage in early ischemic stroke stage. J Neurosci. 2012, 32 (9): 3044-3057.
Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA: Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007, 27 (4): 697-709.
Yang Y, Rosenberg GA: MMP-mediated disruption of claudin-5 in the blood–brain barrier of rat brain after cerebral ischemia. Method Mol Biol. 2011, 762: 333-345.
Li N, Liu YF, Ma L, Worthmann H, Wang YL, Wang YJ, Gao YP, Raab P, Dengler R, Weissenborn K, Zhao XQ: Association of molecular markers with perihematomal edema and clinical outcome in intracerebral hemorrhage. Stroke. 2013, 44 (3): 658-663.
Wang J, Tsirka SE: Neuroprotection by inhibition of matrix metalloproteinases in a mouse model of intracerebral haemorrhage. Brain. 2005, 128 (Pt 7): 1622-1633.
Xue M, Hollenberg MD, Demchuk A, Yong VW: Relative importance of proteinase-activated receptor-1 versus matrix metalloproteinases in intracerebral hemorrhage-mediated neurotoxicity in mice. Stroke. 2009, 40 (6): 2199-2204.
Rosenberg GA, Navratil M: Metalloproteinase inhibition blocks edema in intracerebral hemorrhage in the rat. Neurology. 1997, 48 (4): 921-926.
Wells JE, Biernaskie J, Szymanska A, Larsen PH, Yong VW, Corbett D: Matrix metalloproteinase (MMP)-12 expression has a negative impact on sensorimotor function following intracerebral haemorrhage in mice. Eur J Neurosci. 2005, 21 (1): 187-196.
Grossetete M, Rosenberg GA: Matrix metalloproteinase inhibition facilitates cell death in intracerebral hemorrhage in mouse. J Cereb Blood Flow Metab. 2008, 28 (4): 752-763.
Tang J, Liu J, Zhou C, Alexander JS, Nanda A, Granger DN, Zhang JH: Mmp-9 deficiency enhances collagenase-induced intracerebral hemorrhage and brain injury in mutant mice. J Cereb Blood Flow Metab. 2004, 24 (10): 1133-1145.
Gregson BA, Broderick JP, Auer LM, Batjer H, Chen XC, Juvela S, Morgenstern LB, Pantazis GC, Teernstra OP, Wang WZ, Zuccarello M, Mendelow AD: Individual patient data subgroup meta-analysis of surgery for spontaneous supratentorial intracerebral hemorrhage. Stroke. 2012, 43 (6): 1496-1504.
Mendelow AD, Gregson BA, Rowan EN, Murray GD, Gholkar A, Mitchell PM, Investigators SI: Early surgery versus initial conservative treatment in patients with spontaneous supratentorial lobar intracerebral haematomas (STICH II): a randomised trial. Lancet. 2013, 382 (9890): 397-408.
Morgan T, Zuccarello M, Narayan R, Keyl P, Lane K, Hanley D: Preliminary findings of the minimally-invasive surgery plus rtPA for intracerebral hemorrhage evacuation (MISTIE) clinical trial. Acta Neurochir Suppl. 2008, 105: 147-151.
Mould WA, Carhuapoma JR, Muschelli J, Lane K, Morgan TC, McBee NA, Bistran-Hall AJ, Ullman NL, Vespa P, Martin NA, Awad I, Zuccarello M, Hanley DF: MISTIE Investigators: minimally invasive surgery plus recombinant tissue-type plasminogen activator for intracerebral hemorrhage evacuation decreases perihematomal edema. Stroke. 2013, 44 (3): 627-634.
Keep RF, Xiang J, Ennis SR, Andjelkovic A, Hua Y, Xi G, Hoff JT: Blood–brain barrier function in intracerebral hemorrhage. Acta Neurochir Suppl. 2008, 105: 73-77.
Acknowledgments
This work was supported by the National Institutes of Health grants NS034709, NS073959, NS079157, NS084049, NS075757. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RFK drafted and wrote the manuscipt. NZ, JX, AVA, YH and GX commented on and revised the manuscript. All authors have read and approved the final version of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Keep, R.F., Zhou, N., Xiang, J. et al. Vascular disruption and blood–brain barrier dysfunction in intracerebral hemorrhage. Fluids Barriers CNS 11, 18 (2014). https://doi.org/10.1186/2045-8118-11-18
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2045-8118-11-18