
Abstract
Background
Microdeletions of 14q22q23 have been associated with eye abnormalities and pituitary defects. Other phenotypic features in deletion carriers including hearing loss and response to growth hormone therapy are less well recognized. We studied genotype and phenotype of three newly identified children with 14q22q23 deletions, two girls and one boy with bilateral anophthalmia, and compared them with previously published deletion patients and individuals with intragenic defects in genes residing in the region.
Results
The three deletions were de novo and ranged in size between 5.8 and 8.9 Mb. All three children lacked one copy of the OTX2 gene and in one of them the deletion involved also the BMP4 gene. All three patients presented partial conductive hearing loss which tended to improve with age. Analysis of endocrine and growth phenotypes showed undetectable anterior pituitary, growth hormone deficiency and progressive growth retardation in all three patients. Growth hormone therapy led to partial catch-up growth in two of the three patients but just prevented further height loss in the third.
Conclusions
The pituitary hypoplasia, growth hormone deficiency and growth retardation associated with 14q22q23 microdeletions are very remarkable, and the latter appears to have an atypical response to growth hormone therapy in some of the cases.
Similar content being viewed by others

Background
The morphogenesis of midline brain structures, eyes, optic nerves and optic tracts is governed by a cascade of transcription factors including SOX2, OTX2 and BMP4 [1]. Congenital anophthalmia, which is among the most severe consequences of defects in this cascade, is often accompanied by pituitary dysfunction and growth failure due to growth hormone (GH) deficiency [2–6]. Mutations in SOX2 are the most common cause of anophthalmia, and 10% of their carriers also show growth retardation [1]. The co-occurrence of eye malformations and GH deficiency is the highest (30%) in patients with OTX2 mutations [3]. BMP4 defects can induce a similar brain and ocular phenotype [6, 7]. Mutations ranging from single nucleotide substitutions to cytogenetically visible deletions have been reported in OTX2 and BMP4, which are both located in 14q22q23. We identified three unrelated patients with anophthalmia, partial hearing loss and pituitary defects due to microdeletions of this region. This gave us a unique opportunity to study not only the ocular phenotypes which are well associated with these rare genetic defects, but also other phenotypic features which are less well characterized.
Case presentation
Clinical reports
Patient 1
The girl (Figure 1a) was born to healthy parents of Mali origin at 38 weeks of gestation by Caesarean section because of abnormal fetal cardiac rhythm. Her birth weight, length and head circumference (HC) were 3,110 g (50th centile), 51 cm (75th centile), and 35.5 cm (75th centile), respectively. She showed bilateral anophthalmia. Brain magnetic resonance imaging (MRI) detected hypoplastic orbits, and absent optic nerves and optic chiasm. The anterior pituitary was undetectable, the posterior pituitary was ectopic with hypoplasia of the pituitary stalk, and the sella was small and flat. She had high forehead, microretrognathia, high arched palate, large ears, persistent hypotonia and right postaxial polydactyly. Cardiac examination revealed a systolic murmur and a perimembranous interventricular septal defect of 3 mm. Skeletal radiography and external genitalia were normal.

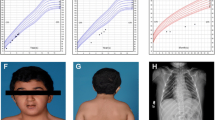
Facial photographs of the patients and their growth characteristics. (a) Patient 1 at the age of 6 months; (b) Patient 2 at the age of 7 months; (c) Patient 3 at the age of 22 months; (d) Development of body length with the onset of GH administration indicated by arrows.
After birth, provoked otoacustic emissions were abnormal on the left side. Auditory evoked potentials at 3 years of age found a 60 dB threshold hearing in both ears. Because of persistent middle ear effusion, tympanostomic tubes were inserted, but had to be removed due to chronic otorrhea.A very low serum IGF-I (9 μg/l) and cortisol (<10 nmol/l) levels at 10 days of life suggested GH deficiency and a cortisol function defect (confirmed by ACTH stimulation test). The levels of other pituitary hormones were normal (TSH 3.48 mUI/l, T4 17.8 pmol/l). The growth of Patient 1 progressively deteriorated from −2.4 SD (60 cm) at 6 months to −3.5 SD (71 cm) at 1.6 years and to −4.9 SD (75.5 cm) at 2.7 years of age (Figure 1d). GH therapy at a dose of 35 μg/kg/day initiated at the age of 2.7 years led to an improved growth rate and stepwise normalization of circulating IGF-I (4, 11 and 135 μg/l at the onset and after 3 and 12 months of therapy, respectively; normal age-specific serum IGF-I range is 51–218 μg/l).
Patient 2
The girl (Figure 1b) was born to healthy Czech parents in the 41st week of gestation with a weight of 3,500 g (50th centile), length of 52 cm (75th centile), and HC of 33 cm (10th centile). Bilateral anophthalmia and relative microcephaly were noted at birth. Brain MRI revealed absence of optic nerves, optic chiasm and optic tracts. The sella was flat, the pituitary stalk and posterior pituitary were present and normally located, but the anterior pituitary was undetectable. The patient showed profound hypotonia and very large, low-set dysplastic ears, high prominent forehead, high frontal hairline, and wide nose with horizontal nostrils, but no cardiac or genital defects. Radiography revealed the presence of 13 pairs of ribs and unpent arcs of vertebral corpus Th1.
Investigation of otoacustic emissions was not successful in the newborn. At 6 months of age she had normal hearing at the left side and moderate conductive hearing loss at the right side. Stenotic Eustachian tubes likely led to decreased pressure in the middle ear cavity. The hearing loss tended to improve with age.Initially, the growth of the patient was normal. However, it started to decelerate to −0.8 SD (70 cm) at 10 months, −1.4 SD (76.3 cm) at 1.4 years and −1.8 SD (79 cm) at 1.8 years of age (Figure 1d). Endocrine assessment revealed GH deficiency (3.53 ug/l following insulin-induced hypoglycemia at 17 months of age) and IGF-I deficiency (11 ug/l; −1.79 SD) but normal other pituitary functions (TSH 1.19 mIU/l, fT4 12.2 pmol/l, FSH 8.9 IU/l, LH 0.9 IU/l, cortisol 555 nmol/l, prolactin 4.7 ug/l). GH therapy at a dose of 25 μg/kg/day was initiated at the age of 1.8 years. It improved the growth rate and the serum IGF-I level (5, 75 and 116 μg/l at the onset, 12 and 24 months of therapy, respectively) but did not lead to catch-up growth (99 cm at 4 years, i.e. -1.5 SD, Figure 1d).
Patient 3
The boy (Figure 1c) was born to healthy Czech parents in the 37th week of gestation by Caesarean section due to intrauterine growth retardation with a weight of 2,060 g and length of 44 cm (both below the 3rd centile for the gestational age), bilateral anophthalmia and marked hypotonia. Brain MRI revealed absent optic chiasm; however, extraocular muscles were preserved. Similarly to Patient 2, the sella was flat, the pituitary stalk and posterior pituitary were present and normally located, but the anterior pituitary was undetectable. Patient 3 showed mesocephaly with prominent narrow forehead, a small narrow face and a wide nasal bridge. He had no apparent morphological ear abnormalities and no cardiac, spinal, abdominal, or genital defects. His bilateral testicular retention required surgical management.
In the neonatal period, the investigation of otoacustic emissions was unsuccessful. At the age of 3 months he showed mild hearing loss of the right ear and medium hearing loss of the left ear. Despite the tympanometry was normal, the hearing loss was apparently conductive due to stenotic ear canals. Also in this patient hearing gradually improved with age.Intrauterine growth retardation was followed by severe postnatal growth failure: at the age of 7 weeks the boy had a length of 48.4 cm (−3.7 SD), and weight of 3,010 g, and his growth had further deteriorated (Figure 1d). He suffered from GH deficiency (1.79 ug/l following clonidine stimulation at 2.3 years of age) and IGF-I deficiency (2 ug/l). Other pituitary functions were apparently normal (TSH 2.75 mIU/l, fT4 11.8 pmol/l, FSH 0.48 IU/l, LH 0.07 IU/l, cortisol 201 nmol/l, prolactin 5.7 ug/l); however, the bilateral testicular retention might be suggestive of a gonadotropin deficiency. GH therapy at a dose of 25 μg/kg/day was initiated at the age of 2.3 years. His height velocity on therapy was atypical, with only a moderate increase within the first year of GH administration but a marked increase thereafter (92.0 cm at 4.1 years, i. e. -3.17 SD, Figure 1d). Serum IGF-I levels were gradually increasing to 6, 19, 33, 56 and 67 μg/l at the onset and during the first two years of therapy.
Laboratory methods
The study was approved by the local ethics committees and all analyses were performed after proper informed consent. Karyotyping of the patients was performed using standard methods. Array comparative genomic hybridization (CGH) used 180K and 105K CGH arrays (Agilent Technologies) in Patient 1 and Patients 2/3, respectively. The deletions were confirmed in the patients and tested in the parents using fluorescence in situ hybridization (FISH) with probes RP11-533L7/RP11-550 M19 and RP11-550M19 (BlueGnome) in Patients 1 and 2, respectively. In Patient 3 the deletion was confirmed using a 60K CGH array (Agilent Technologies).
Results
Karyotyping did not show chromosome abnormalities in any of the three patients. Array CGH revealed interstitial deletions of 14q22q23 of varying size (Figure 2) in the absence of additional relevant submicroscopic aberrations. Patient 1 carried an 8.8 Mb long deletion (chr14:50293781–59068634, hg18) removing multiple genes including BMP4 and OTX2 but not the SIX gene cluster. Patient 2 had a deletion of 8.9 Mb (chr14:54251697–63177878) affecting OTX2 and the SIX gene cluster. Patient 3 had a 5.8 Mb long deletion (chr14:54431790–60167626) removing OTX2 and a part of the SIX gene cluster. Using independent methods, all deletions were confirmed in the patients but not in any of the parents, thus indicating the de novo nature of the aberrations.

Schematic representation of the 14q22q23 region affected by deletions in the patients. The deletions in our patients are shown as thick hatched bars, the deletions overlapping OTX2 and/or BMP4 in published cases as dotted bars. Intragenic deletions in OTX2 or BMP4 are not shown. Protein-coding genes are indicated by black rectangles (OTX2 or BMP4 are in red). The chromosome 14 banding pattern and megabase scale are also included.
Discussion
We present a case series of three patients with bilateral anophthalmia caused by microdeletions of 14q22q23. Their phenotype was further characterized by hearing impairment, abnormal pituitary development leading to GH deficiency and early growth failure, and dysmorphic facial features. The overview of phenotypes observed in published cases with 14q22q23 deletions and in our three patients is in Table 1.
The 14q22q23 region is critical for eye and pituitary development. Anophthalmia and other ocular anomalies were associated with heterozygous defects in OTX2[3, 8, 9] or BMP4[7, 10, 11], and also with deletions involving both these genes [4, 6, 12]. While OTX2 was deleted in all our patients, BMP4 was deleted only in Patient 1 (Figure 2). Nevertheless, the ocular phenotype was similar in all three children. The phenotypic effect of OTX2 and BMP4 disruptions is very variable ranging between anophthalmia/microphthalmia, corneal opacity and no abnormality at all, even in family members with the same mutation [2, 5, 9–13], and the phenotype does not have to be more severe in patients with combined OTX2/BMP4 defects [4, 12]. Patients 2 and 3 also lacked SIX6; however, defects of this candidate gene have not been identified in anophthalmia [1].
All three patients suffered from transient partial conductive hearing loss. Previous reports of patients with 14q22q23 deletions were inconsistent, varying from not mentioning the hearing status over normal function [7, 12] to severe unilateral hearing loss [4, 9], indicating very variable expressivity. We speculate that skeletal abnormalities of the facial-cranial junction of the skull associated with anophthalmia due to the OTX2 defect can induce stenosis of ear canals and/or Eustachian tubes and hearing impairment. Changing proportions of these structures during growth could also explain the gradual improvement of hearing with age observed in our patients. Interestingly, Patient 2 with large low-set dysplastic ears was the only our patient with SIX1 disruption. Homozygous knockout of this gene in mice causes malformations of the auditory system including outer ears [14]. Deletions in two published patients also involved SIX1 and were associated with malformed ears, although differently from Patient 2 [4, 9]. OTX2 defects themselves could also contribute to ear anomalies [15].
Our patients showed abnormal pituitary development, GH deficiency and growth retardation. Normal birth parameters and postnatal growth failure similar to that in Patients 1 and 2 were reported in some cases with 14q22q23 deletions [3, 4, 11, 12]. On the other hand, Patient 3 suffered from severe intrauterine growth retardation without postnatal catch-up. An improvement of growth was evident after GH therapy in all our patients although the response varied. In Patients 1 and 3 the therapy induced an increase of growth velocity and improvement of their height, whereas in Patient 2 it just prevented further growth deterioration. However, in Patient 2 the growth failure was least pronounced with height of −1.8 SD at the start of the GH therapy, and this fact could influence the GH response. Similarly, two published 14q22q23 deletion patients treated with GH remained with their height at −2 SD after five and three years of therapy [4, 13], and the height of a boy with a missense OTX2 mutation remained at −2.3 SD after eight years of therapy [2]. As no reports are currently available on final height of 14q22q23 deletion patients, it remains to be seen if Patients 1 and 3 correct their growth failure completely or if they just reach the current height range of Patient 2. Differences in the deleted genes do not offer an obvious explanation for the differences in responsiveness to the GH therapy. Two genes involved in pituitary development, OTX2 and SIX6, were disrupted in Patients 2 and 3. Patient 1 had a deletion of OTX2 and BMP4, but not of SIX6. The differences in pituitary morphology between Patient 1 and Patients 2 and 3 can also be attributed solely to the variability seen among carriers of isolated OTX2 defects [1, 3].
Finally, polydactyly was present in Patient 1 with a deletion of BMP4. This gene plays an important role in the onset of endochondral bone formation in humans, and its mutations were associated with polysyndactyly [4, 11].
Recently, two papers were published describing patients with deletions overlapping the proximal and distal part of the 14q22q23 region [16, 17]. A family with Frias syndrome carried a deletion of 14q22.1q22.3 spanning the interval between GNG2 and KTN1, with BMP4 haploinsufficiency being likely responsible for the phenotype which included hypoplasia of corpus callosum, minor ocular anomalies, specific tooth defects, digit anomalies, short stature and intellectual impairment [16]. The other deletion involving 14q22.3q23.2 and extending from PSMA3 over the SIX cluster to SYNE2 was identified in a patient with facial dysmorphism, choanal atresia, esophageal reflux, defects of hands and feet, seizures and intellectual disability [17]. Interestingly, in the latter case neither microphthalmia/anophthalmia nor pituitary anomalies were present (MRI was normal), and neither OTX2 nor BMP4 were deleted. These papers further illustrate the intra- and interfamilial variability which, together with biased and incomplete reporting of the phenotypes, complicates the genotype-phenotype correlation in patients with unique deletions affecting multiple genes which participate in different molecular pathways [16, 17].
Conclusions
Our case series study of three patients with deletions of 14q22q23 demonstrated the phenotypic features and variable expressivity of this genetic defect. Comparison with previously published patients with similar microdeletions and mutations in the OTX2 gene suggests that most symptoms presented by affected patients could be attributed to OTX2 haploinsufficiency. Growth retardation due to GH deficiency is very remarkable in these patients and GH treatment can increase the growth velocity especially in the most severe cases but the response can be atypical. The hearing impairment can be transient and improves with age.
Consent
Written informed consent was obtained from the parents of the patients for publication of this report and the accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
References
Slavotinek AM: Eye development genes and known syndromes. Mol Genet Metab 2011, 104: 448–456. 10.1016/j.ymgme.2011.09.029
Ashkenazi-Hoffnung L, Lebenthal Y, Wyatt AW, Ragge NK, Dateki S, Fukami M, Ogata T, Phillip M, Gat-Yablonski G: A novel loss-of-function mutation in OTX2 in a patient with anophthalmia and isolated growth hormone deficiency. Hum Genet 2010, 127: 721–729. 10.1007/s00439-010-0820-9
Dateki S, Kosaka K, Hasegawa K, Tanaka H, Azuma N, Yokoya S, Muroya K, Adachi M, Tajima T, Motomura K, Kinoshita E, Moriuchi H, Sato N, Fukami M, Ogata T: Heterozygous orthodenticle homeobox 2 mutations are associated with variable pituitary phenotype. J Clin Endocrinol Metab 2010, 95: 756–764. 10.1210/jc.2009-1334
Nolen LD, Amor D, Haywood A, St Heaps L, Willcock C, Mihelec M, Tam P, Billson F, Grigg J, Peters G, Jamieson RV: Deletion at 14q22–23 indicates a contiguous gene syndrome comprising anophthalmia, pituitary hypoplasia, and ear anomalies. Am J Med Genet A 2006, 140: 1711–1718.
Schilter KF, Schneider A, Bardakjian T, Soucy JF, Tyler RC, Reis LM, Semina EV: OTX2 microphthalmia syndrome: four novel mutations and delineation of a phenotype. Clin Genet 2011, 79: 158–168. 10.1111/j.1399-0004.2010.01450.x
Bakrania P, Efthymiou M, Klein JC, Salt A, Bunyan DJ, Wyatt A, Ponting CP, Martin A, Williams S, Lindley V, Gilmore J, Restori M, Robson AG, Neveu MM, Holder GE, Collin JR, Robinson DO, Farndon P, Johansen-Berg H, Gerrelli D, Ragge NK: Mutations in BMP4 cause eye, brain, and digit developmental anomalies: overlap between the BMP4 and hedgehog signaling pathways. Am J Hum Genet 2008, 82: 304–319. 10.1016/j.ajhg.2007.09.023
Reis LM, Tyler RC, Schilter KF, Abdul-Rahman O, Innis JW, Kozel BA, Schneider AS, Bardakjian TM, Lose EJ, Martin DM, Broeckel U, Semina EV: BMP4 loss-of-function mutations in developmental eye disorders including SHORT syndrome. Hum Genet 2011, 130: 495–504. 10.1007/s00439-011-0968-y
Wyatt A, Bakrania P, Bunyan DJ, Osborne RJ, Crolla JA, Salt A, Ayuso C, Newbury-Ecob R, Abou-Rayyah Y, Collin JR, Robinson D, Ragge N: Novel heterozygous OTX2 mutations and whole gene deletions in anophthalmia, microphthalmia and coloboma. Hum Mutat 2008, 29: E278-E283. 10.1002/humu.20869
Pearce ZD, Droste PJ, Aaberg TM Jr, Hassan AS: Ophthalmic and systemic findings in interstitial deletions of chromosome 14q: a case report and literature review. Ophthalmic Genet 2012, 33: 161–166. 10.3109/13816810.2012.655359
Lumaka A, Van Hole C, Casteels I, Ortibus E, De Wolf V, Vermeesch JR, Lukusa T, Devriendt K: Variability in expression of a familial 2.79 Mb microdeletion in chromosome 14q22.1–22.2. Am J Med Genet A 2012, 158A: 1381–1387. 10.1002/ajmg.a.35353
Hayashi S, Okamoto N, Makita Y, Hata A, Imoto I, Inazawa J: Heterozygous deletion at 14q22.1-q22.3 including the BMP4 gene in a patient with psychomotor retardation, congenital corneal opacity and feet polysyndactyly. Am J Med Genet A 2008, 146A: 2905–2910. 10.1002/ajmg.a.32519
Takenouchi T, Nishina S, Kosaki R, Torii C, Furukawa R, Takahashi T, Kosaki K: Concurrent deletion of BMP4 and OTX2 genes, two master genes in ophthalmogenesis. Eur J Med Genet 2013, 56: 50–53. 10.1016/j.ejmg.2012.10.007
Delahaye A, Bitoun P, Drunat S, Gerard-Blanluet M, Chassaing N, Toutain A, Verloes A, Gatelais F, Legendre M, Faivre L, Passemard S, Aboura A, Kaltenbach S, Quentin S, Dupont C, Tabet AC, Amselem S, Elion J, Gressens P, Pipiras E, Benzacken B: Genomic imbalances detected by array-CGH in patients with syndromal ocular developmental anomalies. Eur J Hum Genet 2012, 20: 527–533. 10.1038/ejhg.2011.233
Zheng W, Huang L, Wei ZB, Silvius D, Tang B, Xu PX: The role of Six1 in mammalian auditory system development. Development 2003, 130: 3989–4000. 10.1242/dev.00628
Chassaing N, Sorrentino S, Davis EE, Martin-Coignard D, Iacovelli A, Paznekas W, Webb BD, Faye-Petersen O, Encha-Razavi F, Lequeux L, Vigouroux A, Yesilyurt A, Boyadjiev SA, Kayserili H, Loget P, Carles D, Sergi C, Puvabanditsin S, Chen CP, Etchevers HC, Katsanis N, Mercer CL, Calvas P, Jabs EW: OTX2 mutations contribute to the otocephaly-dysgnathia complex. J Med Genet 2012, 49: 373–379. 10.1136/jmedgenet-2012-100892
Martinez-Fernandez ML, Bermejo-Sanchez E, Fernandez B, Macdonald A, Fernandez-Toral J, Martinez-Frias ML: Haploinsufficiency of BMP4 gene may be the underlying cause of Frias syndrome. Am J Med Genet A 2014, 164: 338–345. 10.1002/ajmg.a.36224
Martinez-Frias ML, Ocejo-Vinyals JG, Arteaga R, Martinez-Fernandez ML, Macdonald A, Perez-Belmonte E, Bermejo-Sanchez E, Martinez S: Interstitial deletion 14q22.3-q23.2: genotype-phenotype correlation. Am J Med Genet A 2014. in press
Acknowledgments
We thank the families for their cooperation. This work was supported by Direction de l’Hospitalisation et de l’Organisation des Soins, grants NT13692 and DRO UH Motol 00064203 from the Czech Ministry of Health, and CZ.2.16/3.1.00/24022.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SB, ZSl, PD, ABS, CM, LT and MG carried out the cytogenetic and molecular cytogenetic studies; JL, ZSu, MS, EZ, SB, CM, KM and PL participated in the analysis of the phenotype and genetic counseling; PD, ZSe, JL and SB drafted the manuscript; ZSu edited the manuscript; ZSe, JL and GT conceived, designed and coordinated the study. All authors read and approved the final manuscript.
Sophie Brisset, Zuzana Slamova, Petra Dusatkova contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Brisset, S., Slamova, Z., Dusatkova, P. et al. Anophthalmia, hearing loss, abnormal pituitary development and response to growth hormone therapy in three children with microdeletions of 14q22q23. Mol Cytogenet 7, 17 (2014). https://doi.org/10.1186/1755-8166-7-17
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8166-7-17