
Abstract
Primary sclerosing cholangitis (PSC) is a chronic cholestatic liver disease of unknown aetiology characterised by inflammation and fibrosis of the biliary tree. The mean age at diagnosis is 40 years and men are affected twice as often as women. There is a reported annual incidence of PSC of 0.9–1.31/100,000 and point prevalence of 8.5–13.6/100,000. The onset of PSC is usually insidious and many patients are asymptomatic at diagnosis or have mild symptoms only such as fatigue, abdominal discomfort and pruritus In late stages, splenomegaly and jaundice may be a feature. In most, the disease progresses to cirrhosis and liver failure. Cholangiocarcinoma develops in 8–30% of patients. PSC is thought to be immune mediated and is often associated with inflammatory bowel disease, especially ulcerative colitis. The disease is diagnosed on typical cholangiographic and histological findings and after exclusion of secondary sclerosing cholangitis. Median survival has been estimated to be 12 years from diagnosis in symptomatic patients. Patients who are asymptomatic at diagnosis, the majority of whom will develop progressive disease, have a survival rate greater than 70% at 16 years after diagnosis. Liver transplantation remains the only effective therapeutic option for patients with end-stage liver disease from PSC, although high dose ursodeoxycholic acid may have a beneficial effect.
Similar content being viewed by others
Disease name
Primary sclerosing cholangitis (PSC).
Definition
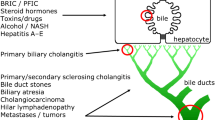
Primary sclerosing cholangitis (PSC) is a chronic cholestatic liver disease of intra and/or extrahepatic bile ducts. A concentric obliterative fibrosis occurs which leads to bile duct strictures, which in turn can progress to biliary cirrhosis and liver failure. Cholangiocarcinoma develops in 8–30% of patients [1, 2].
Secondary sclerosing cholangitis (SSC) is a sclerosing cholangitis that occurs secondary to a known pathogenic process or injury such as obstruction of the bile ducts due to tumour or gallstones; surgical, chemical or ischaemic injury; bacterial infections or congenital abnormalities. SSC has clinical features similar to those of PSC.
Associated diseases
Primary association
Inflammatory bowel disease.
Others
Coeliac disease, diabetes mellitus, rheumatoid arthritis, Sjogren's disease, systemic sclerosis, chronic pancreatitis, cystic fibrosis, retroperitoneal fibrosis, sarcoidosis, thyroiditis, systemic lupus erythematosus (SLE), lupus nephritis, autoimmune haemolytic anaemia, idiopathic thrombocytopenic purpura, Langerhans cell histiocytosis, membranous nephropathy, Peyronie's disease, vasculitis, gallbladder disease.
Complications
Colorectal cancer, cholangiocarcinoma.
Epidemiology
PSC occurs with a 2:1 male predominance. Little is known about the incidence and prevalence of PSC. There is a reported annual incidence of PSC of 0.9–1.31/100,000 and point prevalence of 8.5–13.6/100,000, which appears to be increasing [3–5]. This probably reflects ascertainment bias rather than a true increase. Patients usually present in the third to fifth decade but PSC is recognised as a cause of chronic liver disease in children. It is also unusual to find more than one family member affected.
PSC is frequently associated with inflammatory bowel disease (IBD), especially ulcerative colitis and, less often, with colonic Crohn's disease. Approximately three quarters of Caucasian patients with PSC have IBD [6, 7] and 2–7.5% of patients with ulcerative colitis have PSC [8–10]. This may be an underestimate as patients with PSC may have normal liver function tests. The symptoms of IBD may predate or follow the diagnosis of PSC. The clinical course of ulcerative colitis in PSC patients is often mild and there is a higher prevalence of rectal sparing compared with controls (52% vs 6%). There is also a higher prevalence of backwash ileitis (51% vs 7%) [11]. The risk of colonic dysplasia and colorectal cancer is higher in those with PSC. A long history of IBD is also a risk factor for cholangiocarcinoma development. Colectomy does not alter the course of PSC [12].
Clinical description and diagnostic methods
The diagnosis of PSC is based on a combination of clinical features and cholestatic liver function tests with typical cholangiographic findings and confirmed by characteristic histological abnormalities. In small duct disease, patients have typical biochemical and histological features but a normal cholangiogram. All secondary causes of cholangitis and other causes of liver disease should be excluded.
Clinical features
The clinical course of PSC is highly variable. Patients may present with abnormal liver function tests on a background of IBD. Patients may remain asymptomatic for years or may develop symptoms of fatigue, abdominal discomfort, pruritus and weight loss. Ultimately patients may develop liver failure or cholangiocarcinoma.
Examination may be normal early in disease. As the disease progresses, patients may develop hepatomegaly. In late stages splenomegaly and jaundice may be a feature. Signs of liver failure and portal hypertension are late signs.
Laboratory tests
Alkaline phosphatase is usually raised and transaminases and gamma-glutamyl transpeptidase (GGT) can also be elevated. Patients with PSC often have marked fluctuations in liver function tests and these are not predictive of prognosis [29]. Results of hepatic synthetic function, i.e. serum albumin and prothrombin time (PT), become abnormal with advanced disease.
Some immunological tests may help in the diagnosis of PSC. Hypergammaglobulinaemia occurs in a third of patients and immunoglobulin M (IgM) levels are increased in 50% of patients with advanced disease.
Imaging
Once abnormal biochemistry is revealed on the liver function tests (LFTs), diagnosis is made mainly on the basis of the cholangiogram. A magnetic resonance cholangiogram (MRCP) is being used more frequently for diagnosis and endoscopic retrograde cholangiography (ERCP) is performed more as a therapeutic procedure.
Typical cholangiographic findings show diffuse stricturing of intrahepatic and extrahepatic bile ducts with dilatation of the areas in between (See Figure 1).

Cholangiogram of primary sclerosing cholangitis.
More advanced disease is associated with long strictures. The presence of a dominant stricture should raise the possibility of a cholangiocarcinoma as a complication of PSC. In small duct disease the cholangiogram will be normal. Ultrasound can be helpful in excluding other causes of biliary obstruction such as gallstones and malignancy.
Histology
In early stages, changes associated with PSC can be focal and may be missed on liver biopsy or the changes may be non-specific. The characteristic finding is of concentric "onion-skin" fibrosis surrounding the bile ducts (See Figure 2). Other bile duct abnormalities may include necrosis of epithelial cells, inflammatory infiltrates and fibrosis. There may be intrahepatic bile duct proliferation with ductopaenia or oedema in some portal tracts. In advanced cases, loss of bile ducts can be a feature (vanishing bile duct syndrome). The parenchyma usually shows non-specific changes [30]. Copper storage protein may be seen in advanced cases.
Differential diagnosis
Differential diagnosis should exclude other liver diseases such as autoimmune hepatitis (can coexist with PSC, termed overlap syndrome), primary biliary cirrhosis, viral hepatitis, as well as secondary causes of cholangitis.
Aetiology and pathogenesis
The aetiology and pathogenesis of PSC are still unknown. The following may be important in the development of PSC.
Immunology
Lymphocytic portal tract infiltration and associations with human leukocyte antigen (HLA) haplotypes and autoantibodies are suggestive of an immune mediated basis for this disease. Autoimmune diseases are more frequent in patients with PSC than in patients with IBD who do not have PSC. The most common are diabetes mellitus, coeliac disease and Grave's disease [17]. Rheumatoid arthritis has also been described in association with PSC and may be a clinical marker for those at high risk of rapid progression to cirrhosis [18].
A high prevalence of atypical anti-neutrophil cytoplasmic antibodies (ANCA) are present in the serum in PSC (33–88%). They are also found in ulcerative colitis (60–87%) and type I autoimmune hepatitis (50–96%) [17, 18]. These antibodies are not involved in the pathogenesis of PSC and probably represent an epiphenomenon. Other autoantibodies can be detected in patients with PSC but their presence is not useful for diagnosis or prognosis and, therefore, is of unclear relevance.
Hypergammaglobulinaemia occurs in one third of patients with PSC and IgM levels are increased in 50% of advanced cases [19]. Other immunological changes include a decrease in circulating T cells and an increased ratio of CD4 to CD8 cells.
Grant et al. hypothesised that T Lymphocytes derived from the inflamed gut act as long-lived memory cells [20]. They enter the enterohepatic circulation to gain access to the liver and cause disease when then activated by a certain stimulus [20].
Genetics
PSC is probably acquired through inheriting a combination of genetic polymorphisms that interrelate to cause disease susceptibility. Six different HLA molecules have so far been associated with PSC [21]. Non-MHC (major histocompatibility complex) genes may also play a part but data remains controversial. The importance of genetics in PSC is not fully understood and still being actively researched.
Infections/toxins
The association of PSC and IBD led to Vierling's hypothesis that PSC may be triggered by infections or toxins acting as molecular mimics entering the portal circulation through a permeable colon [22]. The Kupffer cells are activated and cause activated neutrophils, macrophages, lymphocytes and fibroblasts to be attracted, which in turn could then lead to ischaemia, atrophy, cholestasis, fibrosis and then cirrhosis of the biliary cells. However, as PSC can run an independent course from IBD, this suggests bacteraemia on its own is not the cause of PSC development.
Smoking/surgery
There is a well established increased risk of ulcerative colitis in non-smokers when compared to controls. PSC also seems to be a disease of non-smokers, independently of whether the patient has IBD. Previous tonsillectomy may be associated with a decreased risk of PSC. Whilst appendicectomy is not associated with a decreased risk, it may be associated with a delayed onset of PSC. Further studies are required to analyse this association [23–26].
Management including treatment
Since the aetiology and pathogenesis of PSC are still unknown, establishing treatment strategies is difficult. Treatments should ultimately improve symptoms and aim to improve survival. Liver transplant remains the only definitive treatment.
Ursodeoxycholic acid
Ursodeoxycholic acid (UDCA) is a naturally occurring bile acid that improves liver enzyme function in PSC but its effect on liver histology remains controversial. In PSC, there is evidence that high doses (20–30 mg/kg/d) of UDCA may be more effective than moderate doses (10–15 mg/kg/d) at slowing progression of liver fibrosis and cholangiographic appearances [27, 28]. The results of a controlled trial with high dose UDCA for the treatment of PSC has shown a trend towards a prolonged survival in the high dose UDCA group [29]. Studies using immunosuppressant therapy in combination with UDCA show some beneficial effects and need further analysis, as does treatment with antibiotics. In a few patients, features of both PSC and autoimmune hepatitis (AIH) may be present and this is one of the so-called overlap syndromes. This overlap syndrome, in particular, may be more common in children. Immunosuppression appears to be beneficial in this group [30–32].
Dysplasia and cancer
The risk of colorectal dysplasia and colorectal cancer is increased in those with ulcerative colitis who have PSC [33]. Although not evidence-based, current consensus is that these patients should be entered into a surveillance programme and undergo yearly colonoscopies once the diagnosis of PSC has been made. UDCA treatment in patients with PSC and ulcerative colitis has been shown to decrease the risk of colorectal dysplasia and colorectal cancer [34, 35].
Cholangiocarcinoma develops in 8–30% of adult patients with PSC. Hepatocellular carcinoma and gallbladder carcinoma have also been described in PSC. There is also a suggestion of an increased risk of pancreatic carcinoma. Hepatobiliary malignancy is only detected in a proportion of those it is suspected in [36]. Of those PSC patients with cancer, one third will already have hepatobiliary malignancy at the time of or within a year after PSC diagnosis. The risk of hepatobiliary carcinoma is constant after the first year of PSC diagnosis with an incidence rate of 1.5% per year [37]. Measurement and detection of elevated CA19-9 may be a signal to investigate further, but it can also be raised in benign biliary disease [23, 38]. Biliary Mac-2 binding protein has been suggested as a novel diagnostic marker for biliary tract carcinoma, especially when used in combination with biliary CA19-9 levels [39]. Early detection of cholangiocarcinoma is difficult as it may be indistinguishable on cholangiogram from a benign dominant stricture. Brush cytology of dominant strictures has not been shown to have a predictive value [40].
Other complications
Pruritus is a frequent complication and can be disabling. Agents such as cholestyramine (4–16 g/d), rifampicin or naltrexone may all be used [41, 42]. Extra corporeal albumin dialysis has been tried in small numbers of patients with cholestasis, and appears to be an effective alternative for the treatment of patients with pruritus who do not respond to other therapeutic methods [43].
People with advanced PSC are often deficient in the fat-soluble vitamins A, D, E, K and replacement therapy can be given. In osteoporosis, physical exercise and maintenance of adequate calcium and vitamin D may be sufficient in the early stages. Patients with more advanced disease, especially those with IBD, should undergo bone density scanning. Bisphosphonates may be of value.
Recurrent bacterial cholangitis is common, especially in those with strictures, and should be treated with broad-spectrum antibiotics such as ciprofloxacin, which has a high biliary tract penetration. Prophylaxis with oral quinolones may decrease the frequency of cholangitis in those who have a history of recurrent episodes. Cholelithiasis and choledocholithiasis probably occur secondary to chronic cholestasis. ERCP can be used for balloon dilatation or stent placement in the treatment of dominant strictures, and may provide relief of symptoms [44].
Surgery
Liver transplant is an option in the treatment of PSC for those who develop advanced disease. It is difficult to evaluate these patients with regard to timing of transplantation, as disease course is unpredictable. At least 20% of patients undergoing liver transplantation develop recurrent PSC within 5 years [45]. The 5-year survival post transplant is around 75% [46]. The presence of cholangiocarcinoma is traditionally a contraindication to transplant but one study shows a 5 year survival of around 35% for those with cholangiocarcinoma. Thus, if cholangiocarcinoma is only suspected and not definitely diagnosed, the patient should be considered for transplant. Incidental finding of a cholangiocarcinoma during transplant does not affect patient survival significantly [36, 47]. Inflammatory bowel disease symptoms may become more severe post transplantation prompting proctocolectomy, and 5–10% of those with IBD develop colorectal cancer after liver transplant [48].
Prognosis
Median survival has been estimated to be 12 years from diagnosis in symptomatic patients [1, 6, 7]. Patients who are asymptomatic at diagnosis, the majority of whom will develop progressive disease, have a survival rate greater than 70% at 16 years after diagnosis [49].
Patients who have cholestatic liver function tests and a clinical diagnosis of PSC with features consistent with this diagnosis on the liver biopsy, but who have a normal cholangiogram, have small duct PSC; 6–11% of the PSC population have small duct disease. Patients with small duct disease follow a benign course and have a favourable prognosis with regard to survival, transplantation and development of cholangiocarcinoma. The small duct disease does, however, progress in a small number (12%) to large duct disease [50–52].
Unresolved questions
The aetiology of PSC is still unknown.
More needs to be known about the natural history of PSC.
The availability of predictors for development of colonic and bile duct cancer would be useful.

Histological features of primary sclerosing cholangitis.
References
Broome U, Olsson R, Loof L, Bodemar G, Hultcrantz R, Danielsson A, Prytz H, Sandberg-Gertzen H, Wallerstedt S, Lindberg G: Natural history and prognostic factors in 305 Swedish patients with primary sclerosing cholangitis. Gut. 1996, 38: 610-615.
Wiesner RH: Liver transplantation for primary sclerosing cholangitis: timing, outcome, impact of inflammatory bowel disease and recurrence of disease. Best Pract Res Clin Gastroenterol. 2001, 15: 667-680. 10.1053/bega.2001.0212.
Boberg KM, Aadland E, Jahnsen J, Raknerud N, Stiris M, Bell H: Incidence and prevalence of primary biliary cirrhosis, primary sclerosing cholangitis, and autoimmune hepatitis in a Norwegian population. Scand J Gastroenterol. 1998, 33: 99-103. 10.1080/00365529850166284.
Kingham JG, Kochar N, Gravenor MB: Incidence, clinical patterns, and outcomes of primary sclerosing cholangitis in South Wales, United Kingdom. Gastroenterology. 2004, 126: 1929-1930. 10.1053/j.gastro.2004.04.052.
Bambha K, Kim WR, Talwalkar J, Torgerson H, Benson JT, Therneau TM, Loftus EV Jr, Yawn BP, Dickson ER, Melton LJ: Incidence, clinical spectrum, and outcomes of primary sclerosing cholangitis in a United States community. Gastroenterology. 2003, 125: 1364-1369. 10.1016/j.gastro.2003.07.011.
Wiesner RH, Grambsch PM, Dickson ER, Ludwig J, MacCarty RL, Hunter EB, Fleming TR, Fisher LD, Beaver SJ, LaRusso NF: Primary sclerosing cholangitis: natural history, prognostic factors and survival analysis. Hepatology. 1989, 10: 430-436.
Farrant JM, Hayllar KM, Wilkinson ML, Karani J, Portmann BC, Westaby D, Williams R: Natural history and prognostic variables in primary sclerosing cholangitis. Gastroenterology. 1991, 100: 1710-1717.
Olsson R, Danielsson A, Jarnerot G, Lindstrom E, Loof L, Rolny P, Ryden BO, Tysk C, Wallerstedt S: Prevalence of primary sclerosing cholangitis in patients with ulcerative colitis. Gastroenterology. 1991, 100: 1319-1323.
Shepherd HA, Selby WS, Chapman RW, Nolan D, Barbatis C, McGee JO, Jewell DP: Ulcerative colitis and persistent liver dysfunction. Q J Med. 1983, 52: 503-513.
Schrumpf E, Fausa O, Elgjo K, Kolmannskog F: Hepatobiliary complications of inflammatory bowel disease. Semin Liver Dis. 1988, 8: 201-219.
Loftus EV Jr, Harewood GC, Loftus CG, Tremaine WJ, Harmsen WS, Zinsmeister AR, Jewell DA, Sandborn WJ: PSC-IBD: a unique form of inflammatory bowel disease associated with primary sclerosing cholangitis. Gut. 2005, 54: 91-96. 10.1136/gut.2004.046615.
Boberg KM, Bergquist A, Mitchell S, Pares A, Rosina F, Broome U, Chapman R, Fausa O, Egeland T, Rocca G, Schrumpf E: Cholangiocarcinoma in primary sclerosing cholangitis: risk factors and clinical presentation. Scand J Gastroenterol. 2002, 37: 1205-1211. 10.1080/003655202760373434.
Bjornsson E, Kilander A, Olsson RCA: 19-9 and CEA are unreliable markers for cholangiocarcinoma in patients with primary sclerosing cholangitis. Liver. 1999, 19: 501-508.
Ludwig J: Surgical pathology of the syndrome of primary sclerosing cholangitis. Am J Surg Pathol. 1989, 13: 43-49.
Saarinen S, Olerup O, Broome U: Increased frequency of autoimmune diseases in patients with primary sclerosing cholangitis. Am J Gastroenterol. 2000, 95: 3195-3199. 10.1111/j.1572-0241.2000.03292.x.
Gow PJ, Fleming KA, Chapman RW: Primary sclerosing cholangitis associated with rheumatoid arthritis and HLA DR4: is the association a marker of patients with progressive liver disease?. J Hepatol. 2001, 34: 631-635. 10.1016/S0168-8278(00)00060-X.
Lo SK, Fleming KA, Chapman RW: Prevalence of anti-neutrophil antibody in primary sclerosing cholangitis and ulcerative colitis using an alkaline phosphatase technique. Gut. 1992, 33: 1370-1375.
Terjung B, Worman HJ: Anti-neutrophil antibodies in primary sclerosing cholangitis. Best Pract Res Clin Gastroenterol. 2001, 15: 629-642. 10.1053/bega.2001.0209.
Duerr RH, Targan SR, Landers CJ, Sutherland LR, Shanahan F: Anti-neutrophil cytoplasmic antibodies in ulcerative colitis. Comparison with other colitides/diarrheal illnesses. Gastroenterology. 1991, 100: 1590-1596.
Grant AJ, Lalor PF, Salmi M, Jalkanen S, Adams DH: Homing of mucosal lymphocytes to the liver in the pathogenesis of hepatic complications of inflammatory bowel disease. Lancet. 2002, 359: 150-157. 10.1016/S0140-6736(02)07374-9.
Donaldson PT: Genetics of liver disease: immunogenetics and disease pathogenesis. Gut. 2004, 53: 599-608. 10.1136/gut.2003.031732.
Vierling JM: Aetiopathogenesis of primary sclerosing cholangitis. Primary sclerosing cholangitis. Edited by: Manns PCR, Stieihl A, Wiesner R. London: Kluwer Academic Publishers; 1998:9
Mitchell SA, Thyssen M, Orchard TR, Jewell DP, Fleming KA, Chapman RW: Cigarette smoking, appendectomy, and tonsillectomy as risk factors for the development of primary sclerosing cholangitis: a case control study. Gut. 2002, 51: 567-573. 10.1136/gut.51.4.567.
van Erpecum KJ, Smits SJ, van de Meeberg PC, Linn FH, Wolfhagen FH, vanBerge-Henegouwen GP, Algra A: Risk of primary sclerosing cholangitis is associated with nonsmoking behavior. Gastroenterology. 1996, 110: 1503-1506. 10.1053/gast.1996.v110.pm8613056.
Loftus EV Jr, Sandborn WJ, Tremaine WJ, Mahoney DW, Zinsmeister AR, Offord KP, Melton LJ: Primary sclerosing cholangitis is associated with nonsmoking: a case-control study. Gastroenterology. 1996, 110: 1496-502. 10.1053/gast.1996.v110.pm8613055.
Florin TH, Pandeya N, Radford-Smith GL: Epidemiology of appendicectomy in primary sclerosing cholangitis and ulcerative colitis: its influence on the clinical behaviour of these diseases. Gut. 2004, 53: 973-979. 10.1136/gut.2003.036483.
Mitchell SA, Bansi DS, Hunt N, Von Bergmann K, Fleming KA, Chapman RW: A preliminary trial of high-dose ursodeoxycholic acid in primary sclerosing cholangitis. Gastroenterology. 2001, 121: 900-907. 10.1053/gast.2001.27965.
Harnois DM, Angulo P, Jorgensen RA, Larusso NF, Lindor KD: High-dose ursodeoxycholic acid as a therapy for patients with primary sclerosing cholangitis. Am J Gastroenterology. 2001, 96: 1558-1562. 10.1111/j.1572-0241.2001.03777.x.
Olsson R, Boberg KM, de Muckadell OS, Lindgren S, Hultcrantz R, Folvik G, Bell H, Gangsoy-Kristiansen M, Matre J, Rydning A, Wikman O, Danielsson A, Sandberg-Gertzen H, Ung KA, Eriksson A, Loof L, Prytz H, Marschall HU, Broome U: High-dose ursodeoxycholic acid in primary sclerosing cholangitis: a 5-year multicenter, randomized, controlled study. Gastroenterology. 2005, 129: 1464-1472. 10.1053/j.gastro.2005.08.017.
Wilschanski M, Chait P, Wade JA, Davis L, Corey M, St Louis P, Griffiths AM, Blendis LM, Moroz SP, Scully L, et al: Primary sclerosing cholangitis in 32 children: clinical, laboratory, and radiographic features, with survival analysis. Hepatology. 1995, 22: 1415-1422. 10.1016/0270-9139(95)90146-9.
McNair AN, Moloney M, Portmann BC, Williams R, McFarlane IG: Autoimmune hepatitis overlapping with primary sclerosing cholangitis in five cases. Am J Gastroenterol. 1998, 93: 777-784. 10.1111/j.1572-0241.1998.224_a.x.
Gregorio GV, Portmann B, Karani J, Harrison P, Donaldson PT, Vergani D, Mieli-Vergani G: Autoimmune hepatitis/sclerosing cholangitis overlap syndrome in childhood: a 16-year prospective study. Hepatology. 2001, 33: 544-553. 10.1053/jhep.2001.22131.
Kornfeld D, Ekbom A, Ihre T: Is there an excess risk for colorectal cancer in patients with ulcerative colitis and concomitant primary sclerosing cholangitis? A population based study. Gut. 1997, 41: 522-525.
Pardi DS, Loftus EV Jr, Kremers WK, Keach J, Lindor KD: Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology. 2003, 124: 889-893. 10.1053/gast.2003.50156.
Tung BY, Emond MJ, Haggitt RC, Bronner MP, Kimmey MB, Kowdley KV, Brentnall TA: Ursodiol use is associated with lower prevalence of colonic neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Ann Intern Med. 2001, 134: 89-95.
Brandsaeter B, Isoniemi H, Broome U, Olausson M, Backman L, Hansen B, Schrumpf E, Oksanen A, Ericzon BG, Hockerstedt K, Makisalo H, Kirkegaard P, Friman S, Bjoro K: Liver transplantation for primary sclerosing cholangitis; predictors and consequences of hepatobiliary malignancy. J Hepatol. 2004, 40: 815-822. 10.1016/j.jhep.2004.01.002.
Bergquist A, Ekbom A, Olsson R, Kornfeldt D, Loof L, Danielsson A, Hultcrantz R, Lindgren S, Prytz H, Sandberg-Gertzen H, Almer S, Granath F, Broome U: Hepatic and extrahepatic malignancies in primary sclerosing cholangitis. J Hepatol. 2002, 36: 321-327. 10.1016/S0168-8278(01)00288-4.
Lindberg B, Arnelo U, Bergquist A, Thorne A, Hjerpe A, Granqvist S, Hansson LO, Tribukait B, Persson B, Broome U: Diagnosis of biliary strictures in conjunction with endoscopic retrograde cholangiopancreaticography, with special reference to patients with primary sclerosing cholangitis. Endoscopy. 2002, 34: 909-916. 10.1055/s-2002-35298.
Koopmann J, Thuluvath PJ, Zahurak ML, Kristiansen TZ, Pandey A, Schulick R, Argani P, Hidalgo M, Iacobelli S, Goggins M, Maitra A: Mac-2-binding protein is a diagnostic marker for biliary tract carcinoma. Cancer. 2004, 101: 1609-1615. 10.1002/cncr.20469.
Ponsioen CY, Vrouenraets SM, van Milligen de Wit AW, Sturm P, Tascilar M, Offerhaus GJ, Prins M, Huibregtse K, Tytgat GN: Value of brush cytology for dominant strictures in primary sclerosing cholangitis. Endoscopy. 1999, 31: 305-309. 10.1055/s-1999-18.
Wolfhagen FH, Sternieri E, Hop WC, Vitale G, Bertolotti M, Van Buuren HR: Oral naltrexone treatment for cholestatic pruritus: a double-blind, placebo-controlled study. Gastroenterology. 1997, 113: 1264-1269. 10.1053/gast.1997.v113.pm9322521.
Gillespie DA, Vickers CR: Pruritus and cholestasis: therapeutic options. J Gastroenterol Hepatol. 1993, 8: 168-173.
Pares A, Cisneros L, Salmeron JM, Caballeria L, Mas A, Torras A, Rodes J: Extracorporeal albumin dialysis: a procedure for prolonged relief of intractable pruritus in patients with primary biliary cirrhosis. Am J Gastroenterol. 2004, 99: 1105-1110. 10.1111/j.1572-0241.2004.30204.x.
Bjornsson E, Lindqvist-Ottosson J, Asztely M, Olsson R: Dominant strictures in patients with primary sclerosing cholangitis. Am J Gastroenterol. 2004, 99: 502-508. 10.1111/j.1572-0241.2004.04106.x.
Graziadei IW, Wiesner RH, Marotta PJ, Porayko MK, Hay JE, Charlton MR, Poterucha JJ, Rosen CB, Gores GJ, LaRusso NF, Krom RA: Long-term results of patients undergoing liver transplantation for primary sclerosing cholangitis. Hepatology. 1999, 30: 1121-1127. 10.1002/hep.510300501.
Brandsaeter B, Friman S, Broome U, Isoniemi H, Olausson M, Backman L, Hansen B, Schrumpf E, Oksanen A, Ericzon BG, Hockerstedt K, Makisalo H, Kirkegaard P, Bjoro K: Outcome following liver transplantation for primary sclerosing cholangitis in the Nordic countries. Scand J Gastroenterol. 2003, 38: 1176-1183. 10.1080/00365520310006009.
Goss JA, Shackleton CR, Farmer DG, Arnaout WS, Seu P, Markowitz JS, Martin P, Stribling RJ, Goldstein LI, Busuttil RW: Orthotopic liver transplantation for primary sclerosing cholangitis. A 12-year single center experience. Ann Surg. 1997, 225: 472-481. 10.1097/00000658-199705000-00004.
Macfaul GR, Chapman RW: Sclerosing cholangitis. Curr Opin Gastroenterol. 2004, 20: 275-280. 10.1097/00001574-200405000-00013.
Porayko MK, Wiesner RH, LaRusso NF, Ludwig J, MacCarty RL, Steiner BL, Twomey CK, Zinsmeister AR: Patients with asymptomatic primary sclerosing cholangitis frequently have progressive disease. Gastroenterology. 1990, 98: 1594-1602.
Boberg KM, Schrumpf E, Fausa O, Elgjo K, Kolmannskog F, Haaland T, Holter E: Hepatobiliary disease in ulcerative colitis. An analysis of 18 patients with hepatobiliary lesions classified as small-duct primary sclerosing cholangitis. Scand J Gastroenterol. 1994, 29: 744-752.
Angulo P, Maor-Kendler Y, Lindor KD: Small-duct primary sclerosing cholangitis: a long-term follow-up study. Hepatology. 2002, 5: 1494-1500. 10.1053/jhep.2002.33202.
Bjornsson E, Boberg KM, Cullen S, Fleming K, Clausen OP, Fausa O, Schrumpf E, Chapman RW: Patients with small duct primary sclerosing cholangitis have a favourable long term prognosis. Gut. 2002, 51: 731-735. 10.1136/gut.51.5.731.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Worthington, J., Chapman, R. Primary sclerosing cholangitis. Orphanet J Rare Dis 1, 41 (2006). https://doi.org/10.1186/1750-1172-1-41
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1750-1172-1-41