
Abstract
Background
MYB is the largest plant transcription factor gene family playing vital roles in plant growth and development. However, it has not been systematically studied in Salvia miltiorrhiza, an economically important medicinal plant.
Results
Here we report the genome-wide identification and characterization of 110 R2R3-MYBs, the largest subfamily of MYBs in S. miltiorrhiza. The MYB domain and other motifs of SmMYBs are largely conserved with Arabidopsis AtMYBs, whereas the divergence of SmMYBs and AtMYBs also exists, suggesting the conservation and diversity of plant MYBs. SmMYBs and AtMYBs may be classified into 37 subgroups, of which 31 include proteins from S. miltiorrhiza and Arabidopsis, whereas 6 are specific to a species, indicating that the majority of MYBs play conserved roles, while others may exhibit species-specialized functions. SmMYBs are differentially expressed in various tissues of S. miltiorrhiza. The expression profiles are largely consistent with known functions of their Arabidopsis counterparts. The expression of a subset of SmMYBs is regulated by microRNAs, such as miR159, miR319, miR828 and miR858. Based on functional conservation of MYBs in a subgroup, SmMYBs potentially involved in the biosynthesis of bioactive compounds were identified.
Conclusions
A total of 110 R2R3-MYBs were identified and analyzed. The results suggest the complexity of MYB-mediated regulatory networks in S. miltiorrhiza and provide a foundation for understanding the regulatory mechanism of SmMYBs.
Similar content being viewed by others


Background
Salvia is characterized with only two stamens connected to form a lever. It includes about 900 species and is the largest genus of the Labiatae family. Salvia, together with the genera Lepechinia, Melissa, Dorystaechas, Meriandra, Zhumeria, Perovskia and Rosmarinus, forms a monophylectic lineage within the Labiatae [1, 2]. S. miltiorrhiza Bunge, known as Danshen in Chinese, is an economically important medicinal plant species of the Salvia genus. It shows close phylogenetic relationships with other Asian and Mediterranean species in the Salvia genus, such as S. roborowskii and S. glutinosa[2]. S. miltiorrhiza has been widely and successfully used in traditional Chinese medicine (TCM) for hundred of years to treat numerous diseases, such as coronary heart diseases, dysmenorrheal, amenorrhoea, and inflammatory diseases [3, 4]. The main bioactive components of S. miltiorrhiza may be divided into two groups. The first group is abietane type-diterpene quinine pigments, known as tanshinones, which are lipophilic and consist of more than thirty compounds [5]. The second group is hydrophilic phenolic acids, including rosmarinic acid, salvianolic acid A, salvianolic acid B, lithospermic acid, and many other chemicals [6]. Biosynthesis of main bioactive components in S. miltiorrhiza requires the coordination of a series of key enzymes [7]. The expression of genes encoding these key enzymes is regulated by various transcription factors, of which MYBs appear to play significant roles [8, 9]. Identification and characterization of MYB genes in S. miltiorrhiza is very important in understanding the regulatory mechanism of bioactive component biosynthesis. Since MYBs are also vital regulators in plant development and plant responses to various biotic and abiotic stresses, elucidation of MYB-associated regulatory networks may greatly help in improving the growth and defense abilities of S. miltiorrhiza through genetic engineering approaches.
MYB proteins, characterized by the MYB domain, have been widely found in many organisms, including animals, fungi and plants. The MYB domain of MYB proteins is deeply conserved and contains up to four imperfect repeats (R) of about 52 amino acids [10]. Many vertebrates contain three MYB genes, known as c-MYB, A-MYB and B-MYB[11]. The repeats in c-MYB proteins are referred to as R1, R2 and R3. Repeats from other proteins are named based on their similarity to R1, R2 or R3 of c-MYB proteins [10, 12]. In plants, MYB is the largest transcription factor family and may be classified into four subfamilies based on the number of adjacent imperfect repeats [10, 13]. It includes R1R2R3R1/2-MYBs (4R-MYBs), R1R2R3-MYBs (3R-MYBs), R2R3-MYBs and MYBs with a single or partial MYB repeat (1R-MYB or MYB-related), of which R2R3-MYB is the largest subfamily of plant MYBs [10]. The number of R2R3-MYB genes in Arabidopsis thaliana, Populus trichocarpa and Oryza sativa is 137, 198, and 95, respectively [13].
R2R3-MYB proteins contain two repeats similar to R2 and R3 of c-MYB proteins and have been proposed to be evolved from a 3R-MYB gene ancestor by the loss of R1 [14]. In addition to the MYB domain, other less conserved motifs were also found in R2R3-MYB proteins. Based on the similarity of these motifs or phylogenetic relationships of MYB amino acids, R2R3-MYBs may be divided into subgroups. For instance, the large Arabidopsis R2R3-MYB subfamily has been divided into 25 subgroups according to the motifs [10, 12]. However, based on phylogenetic relationships, the number of R2R3-MYB subgroups in Arabidopsis, P. trichocarpa, soybean and maize is 40, 42, 48, and 18, respectively [15–17]. It suggests that the resulting number of R2R3-MYB subgroups in Arabidopsis significantly varies between two approaches used and shows that some subgroups are not deeply conserved among different plant species. Proteins in non-conserved subgroups might have specialized roles.
The functions of numerous R2R3-MYBs have been characterized in Arabidopsis. The known functions were well-summarized into four plant-specific processes, including primary and secondary metabolism, cell fate and identity, developmental processes and responses to biotic and abiotic stresses [10]. It clearly demonstrates functional divergence and significance of Arabidopsis R2R3-MYBs. Extensive studies on the MYB gene family have been conducted in various plant species, such as rice, maize, wheat, poplar, eucalyptus and gentian [18–23]. The results showed that the functions of R2R3-MYBs belonging to the same subgroup were usually conserved among different plant species. For instance, subgroup 7 MYBs, including Arabidopsis AtMYB11, AtMYB12 and AtMYB111 [24], grape VvMYBF1 [25], tomato SlMYB12 [26], gentian GtMYBP3 and GtMYBP4 [27], and many others, are involved in the control of flavonol biosynthesis. Subgroup 6 MYBs, such as Arabidopsis AtMYB75/PAP1, AtMYB90/PAP2, AtMYB113 and AtMYB114 [28], cauliflower BoMYB2 [29], apple MYB110a [30], and pear PyMYB10 [31], regulate anthocyanin biosynthesis. It suggests that the functions of MYBs may be predicted by phylogenetic analysis. So far, little is known about MYBs in S. miltiorrhiza.
S. miltiorrhiza is emerging as a model plant for TCM studies, since its relatively small genome size (~600 Mb), short life cycle, undemanding growth requirements, and significant medicinal value [7]. Recently, the genome of S. miltiorrhiza has been sequenced and a working draft of the genome has been assembled (Chen et al., unpublished). In order to elucidate the role of MYB genes in S. miltiorrhiza development, defense and bioactive component biosynthesis, we performed a genome-wide analysis of the R2R3-MYB gene subfamily in S. miltiorrhiza.
Results and discussion
Identification of 110 S. miltiorrhizaR2R3-MYB genes
Through BLAST analysis of 125 A. thaliana R2R3-MYBs [12] against the current assembly of the S. miltiorrhiza genome (Chen et al., unpublished) and subsequent gene prediction of the retrieved genomic DNA sequences, a total of 110 full-length or near full-length members of the S. miltiorrhiza R2R3-MYB gene subfamily were predicted. Notably, it may not be a complete set of R2R3-MYB genes in S. miltiorrhiza, since the current assembly of the S. miltiorrhiza genome is just a working draft and we do identified from the current assembly some partial sequences with high homology to known R2R3-MYBs in other plant species (data not shown). To verify the results from computational prediction,primers were designed for PCR-amplification of full-length coding sequences (CDSs) of 110 S. miltiorrhiza R2R3-MYBs. A total of 109 CDSs were obtained. Sequence comparison showed that the cloned CDSs of 102 R2R3-MYBs were identical to the predicted sequences. The other seven cloned CDSs, including those of SmMYB1, SmMYB13, SmMYB16, SmMYB35, SmMYB62, SmMYB63 and SmMYB79, differed from the predicted ones by one or a few bases only; However, the deduced amino acids were identical. The results confirm the correctness of MYB gene annotation and suggest the reliability of DNA sequence data. We were not able to clone the CDS of SmMYB108. It could be due to low expression level in the tissues analyzed. All 109 cloned CDSs and the predicted SmMYB108 have been submitted to GenBank. The accession numbers in GenBank are shown in Additional file 1: Table S1.
Phylogenetic analysis of R2R3-MYB proteins from S. miltiorrhiza and Arabidopsis
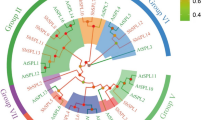
In order to know the relationship of R2R3-MYBs in S. miltiorrhiza and Arabidopsis, a neighbor-joining (NJ) phylogenetic tree was constructed using MEGA4.0 (Figure 1). The results showed that many S. miltiorrhiza MYBs were highly similar to their counterparts in Arabidopsis. Based on the phylogenetic tree and previous results from Arabidopsis[10], R2R3-MYBs in S. miltiorrhiza and Arabidopsis might be classified into 37 subgroups (named S1–S37), of which S1–S25 were named as previously described [10]; while the others were novel. Thirty one of 34 subgroups included proteins from S. miltiorrhiza and Arabidopsis, whereas the other six were specific to S. miltiorrhiza (S29 and S36) or Arabidopsis (S10, S12, S35 and S37). Species-specific subgroups of R2R3-MYBs have also been found in other plant species, such as rice [16], maize [17] and wheat [32]. Since MYBs in a subgroup usually play similar roles or function in a metabolic pathway [10], our results indicate that some MYBs play deeply conserved roles in S. miltiorrhiza and Arabidopsis, while the others may exhibit species-specialized functions.

Phylogenetic relationships of MYB proteins from S. miltiorrhiza and Arabidopsis. Subgroups are highlighted.
Conservation and divergence of MYB domain
The MYB domain of MYB proteins is highly conserved in plants. It contains up to four imperfect repeats (R) of about 52 amino acids [10]. R2R3-MYBs are characterized with two repeats, known as R2 and R3. Consistently, the MYB domain of S. miltiorrhiza and Arabidopsis R2R3-MYBs contain 107 residues, of which 54 form R2, while the other 53 constitute R3 (Figure 2). In order to elucidate sequence features of MYB domain and the degree of conservation of each residue, multiple sequence alignment was performed and sequence logos were created for R2 and R3 of R2R3-MYBs from S. miltiorrhiza and Arabidopsis (Figure 2). The results showed that the distribution of residues in R2 and R3 of S. miltiorrhiza R2R3-MYBs was quite similar to Arabidopsis (Figure 2B and Figure 2D). R2 of both S. miltiorrhiza and Arabidopsis R2R3-MYBs contains three highly conserved tryptophan residues (W) at positions 5, 26 and 46 (Figure 2A and Figure 2B), which may form a tryptophan cluster in the 3-dimensional HTH structure and play significant roles in MYB-DNA interaction [15, 17]. Similarly, three regularly spaced and highly conserved residues, including a phenylalanine (F) and two tryptophan residues, exist at positions 5, 24 and 43 of R3 (Figure 2C and Figure 2D). The highly conserved tryptophan residues were also found in R2R3-MYBs from other plant species, such as Populus trichocarpa[15] and soybean [16]. Although the phenylalanine at position 5 of R3 is conserved in R2R3-MYBs from S. miltiorrhiza, Arabidopsis, and other plant species, such as P. trichocarpa[15] and soybean [16], the degree of conservation is apparently less compared with the tryptophan residues at positions 24 and 43 (Figure 2C and Figure 2D). The functional significance of phenylalanine at position 5 of R3 remains to be elucidated. The other conserved residues in R2 and R3 were mainly distributed between the second and the third residues of three conserved residues described above (Figure 2). The residues between the first and the second conserved tryptophan in R2 and those between the conserved phenylalanine and the first conserved tryptophan in R3, particularly the residues forming the first helix in each repeat, are apparently less conserved (Figure 2). The 3’ region of R2 in both S. miltiorrhiza and Arabidopsis R2R3-MYBs contains a highly conserved LRPD motif (LRPD), which was also observed in P. trichocarpa[15], soybean [16] and maize [17]. The results suggest the conservation of amino acid distribution in the MYB domain of plant R2R3-MYBs. On the other hand, the patterns at positions 15, 22–25, 28 and 36 of R2 and 26, 29, 30 and 52 of R3 are different between S. miltiorrhiza and Arabidopsis (Figure 2), showing divergence of the MYB domain.

Comparison of R2 and R3 sequences in R2R3-MYBs from S. miltiorrhiza and Arabidopsis. (A) and (B) Sequence logos of R2 in MYBs from S. miltiorrhiza (A) and Arabidopsis (B). (C) and (D) Sequence logos of R3 in MYBs from S. miltiorrhiza (C) and Arabidopsis (D). Bits represent the conservation of sequence at a position. The positions with different patterns between S. miltiorrhiza and Arabidopsis are indicated by triangles. Highly conserved tryptophan (W) and phenylalanine (F) residues are indicated by asterisks.
Analysis of conserved motifs other than the MYB domain
It has been shown that the C-terminal region next to the MYB domain of R2R3-MYBs usually contains functionally important motifs, although these motifs are less conserved compared with the MYB domain [10, 12]. Using the MEME suite, a total of 40 motifs were identified in the downstream of MYB domain of S. miltiorrhiza and Arabidopsis R2R3-MYBs (Figure 3 and Additional file 2: Figure S1). The length of motifs varies from 8 to 107 amino acids and the number of motifs in each MYB varies between 0 and 6. No motifs were predicted for 14 SmMYBs and 10 AtMYBs. Although the majority of 40 motifs exist in both S. miltiorrhiza and Arabidopsis R2R3-MYBs, five (motifs 11, 22, 23, 36 and 38) are AtMYB-specific (Figure 4). No SmMYB-specific motifs were identified. Among 40 motifs, motif 1 is the most common motif, which was found in 25 AtMYBs and 23 SmMYBs. The next common motifs are motifs 26 and 32 present in 17 AtMYBs/14 SmMYBs and 18 AtMYBs/12 SmMYBs, respectively. Many S. miltiorrhiza R2R3-MYBs in a subgroup share at least a motif. Consistently, many Arabidoopsis R2R3-MYBs in a subgroup contain same motif(s) as their S. miltiorrhiza orthologues in the subgroup (Additional file 2: Figure S1) [10, 12]. It suggests the conservation of motifs in S. miltiorrhiza and Arabidopsis R2R3-MYBs belonging to a subgroup. The majority of motifs were found in more than one subgroup of R2R3-MYBs, except motifs 8 and 29 existing in S1 and motif 18 in S18. For instance, motifs 21 and 26 widely exist in R2R3-MYBs belonging to 16 and 15 subgroups, respectively (Additional file 2: Figure S1). Taken together, the results suggest that these motifs are evolutionarily conserved and functionally important; however, it is currently unknown for the underlying mechanism of motifs to be under selection and conserved among divergent species.

The number of S. miltiorrhiza and Arabidopsis R2R3- MYB proteins containing conserved motifs outside the MYB domain. Five AtMYB-specific motifs are indicated by triangles.

Clustering of differentially expressed MYB genes in roots (Rt), stems (St), leaves (Le) and flowers (Fl) of S. miltiorrhiza. (A) Ten SmMYBs predominantly expressed in roots. (B) Twelve SmMYBs predominantly expressed in stems. (C) Thirty one SmMYBs predominantly expressed in leaves. (D) Seventeen SmMYBs predominantly expressed in flowers. (E) Thirty nine SmMYBs highly expressed in at least two tissues analyzed. The expression level of each gene is relative to SmUBQ10*10000.
Expression profiling of S. miltiorrhiza R2R3-MYBgenes
In order to elucidate possible roles of R2R3-MYBs in the growth and development of S. miltiorrhiza, we investigated the relative expression level of 110 SmMYBs in roots, stems, leaves and flowers of two-year-old, field nursery-grown S. miltiorrhiza plants using the quantitative real-time RT-PCR method. Transcripts were detected for 109 of 110 SmMYBs (Figure 4 and Additional file 3: Figure S2). The expression of SmMYB108 was undetected, which is consistent with the result from CDS cloning. It indicates that SmMYB108 may be pseudogenes or expressed at specific developmental stages or under special conditions. Of the 109 detectable SmMYBs, 31 (28%) showed predominant expression in leaves, 17 (15.5%) in flowers, 12 (11%) in stems, and 10 (9%) in roots. The other 39 (35.5%) were highly expressed in at least two tissues analyzed (Figure 5). Differential expression of SmMYBs is consistent with the fact that each MYB is usually involved in a limited number of cellular processes.

SmMYBs targeted by miR858, miR828 and miR159/319. (A) miRNA-targeted SmMYB sequences and their locations. (B) Proportion of target sites located in R2, R3 and the divergent region.
Further examination of the expression of SmMYBs in each subgroup showed that some MYB genes in a subgroup shared similar expression profiles and the profiles were consistent with known functions of their Arabidopsis counterparts. For instance, S18 contains 4 SmMYBs and 7 AtMYBs (Figure 2), of which AtMYB33 and AtMYB65 are gibberellin-regulated MYBs (GAMYBs) involved in gibberellin signaling [33–41]. It has been shown that AtMYB33 and AtMYB65 redundantly facilitate anther development. In the anther of a myb33 myb65 double mutant, the tapetum underwent hypertrophy at the pollen mother cell stage [41]. Analyzing the expression of AtMYB33 and AtMYB65 showed complete repression in vegetative tissues through a miR159-associated regulatory mechanism [42]. Consistently, SmMYB62, SmMYB78, SmMYB80 and SmMYB99, four S. miltiorrhiza MYBs include in S18, were predominantly expressed in flowers (Additional file 3: Figure S2), suggesting their significant roles in flower development. In addition, SmMYB37 belonging to S3 was predominately expressed in S. miltiorrhiza stems with plenty of lignin (Additional file 3: Figure S2). It is consistent with the function of AtMYB58 and AtMYB63 belonging to the same subgroup of SmMYB37 in activation of PAL, C4H and 4CL genes involved in the phenylpropanoid pathway and various other genes related to lignin biosynthesis [43].
On the other hand, many SmMYB genes in a subgroup showed differential expression in S. miltiorrhiza. Even so, the expression profiles were still consistent with known functions of their Arabidopsis counterparts. S14 includes 9 SmMYBs and 6 AtMYBs, of which SmMYB47 and AtMYB36 were root-specific. SmMYB100 and AtMYB68 was exclusively found in roots and exhibited low level in flowers. AtMYB37 was predominantly expressed in roots and was also found in flowers and shoots. SmMYB18 was highly expressed in stems and less in leaves. SmMYB60 was highly expressed in leaves and less in stems. SmMYB27 was abundant in roots and less in stems and leaves, while SmMYB8, SmMYB10, SmMYB83, SmMYB89, AtMYB38, AtMYB84 and AtMYB87 were expressed in all tissues analyzed (Additional file 3: Figure S2) [44]. Consistently, AtMYB37, AtMYB38 and AtMYB84 redundantly regulate axillary meristem formation at different stages of vegetative development [45, 46], whereas AtMYB68 is a root growth-specific regulator [46]. AtMYB16 and AtMYB106 belonging to S9 are MIXTA-like transcription factors involved in trichome differentiation, petal cell development and petal morphogenesis [47–52]. Consistently, all S9 SmMYBs, including SmMYB21, SmMYB39, SmMYB51, SmMYB67, SmMYB71 and SmMYB86, were highly expressed in leaves with abundant of trichomes (Additional file 3: Figure S2). In addition, SmMYB39, SmMYB67 and SmMYB71 were also highly expressed in flowers (Additional file 3: Figure S2). Taken together, the results strongly suggest that many SmMYBs play similar roles as AtMYBs in the same subgroup.
miRNA-mediated post-transcriptional regulation of SmMYBs
Plant microRNAs (miRNAs) are a class of endogenous noncoding RNAs with size about 21 nucleotides. They play vital roles in plant development and stress responses mainly through direct cleavage of target mRNAs with perfect or near-perfect complementarities [53–58]. It allows an effective prediction of target sequence through computational approaches [59]. So far, a total of 190 Arabidopsis miRNA gene families, representing 338 mature miRNAs, have been identified [60]. Among them, eight (miR159a, miR159b, miR159c, miR319a, miR319b, miR319c, miR828 and miR858) produced from three gene families, including MIR159/319, MIR828 and MIR858, were found to regulate the expression of various AtMYB genes [10, 13, 61, 62]. The regulatory role of miRNAs in MYBs also exists in apple [63]. In order to elucidate miRNA-mediated posttranscriptional regulation of S. miltiorrhiza MYBs, we searched all SmMYBs for potential targets of deeply conserved miR159, miR319, miR828 and miR858 [64]. A total of 38 SmMYBs with complementary sequence to the member of MIR159/319, MIR828 and MIR858 families were identified (Figure 5). Of 38 SmMYBs, 31 were predicted targets of miR858, 2 were of miR828, 2 were of miR159, while the other 3 were common targets of miR828 and miR858, miR159 and miR319, or miR159, miR319 and miR858. It suggests the identification of total 42 miRNA target sites, of which 36 (85.7%) locate in R3, 1 in R2, and 5 (11.9%) in other regions (Figure 5). The large proportion of putative miRNA target sites in R3 could be due to the deep conservation of sequence in this region. Although all target sites of miR828 and miR858 locate in R3, miR858 target to more conserved 5’ end, while miR828 targets to less conserved 3’ end. It results in that the number of miR858 targets, 33, is much greater than that of miR828, 3. One of four target sites of miR159 locates in R2. The other three target sites of miR159 and two target sites of miR319, including the overlapping target sites of miR159 and miR319 in SmMYB78 and SmMYB80, locate in less conserved regions other than R2 and R3 (Figure 5). It suggests the conservation and divergence of miRNA target sites and indicates that the number of miRNA targets in a gene family is associated with the location of target sites.
The majority of 38 SmMYBs are regulated by a miRNA family, whereas the other two, including SmMYB2 and SmMYB80, are targeted by miRNAs from two families (Figure 1). SmMYB2, which is included in S15 and highly expressed in leaves, is a target of the MIR828 and MIR858 family members. In S15, there are 4 Arabidopsis MYBs, of which AtMYB0, AtMYB23 and AtMYB66 are involved in trichome development [41, 65]; however they are not targets of miR828 and miR858. The other S15 Arabidopsis MYB, known as AtMYB82, is a potential target of miR828 and miR858. Since functions of AtMYB82 are currently unknown, the significance of miR828- and miR858-involved co-regulation remains to be elucidated. Similarly, SmMYB80 belonging to S18 and predominantly expressed in flowers is co-regulated by members of MIR159/319 and MIR858. Further analyzing the regulation role of miRNAs in 4 SmMYBs and 7 AtMYBs included in S18 showed that all of them could be targets of miRNAs. Of 11 MYBs, SmMYB99 and AtMYB101 are regulated by miR159. SmMYB62 and AtMYB81 are targets of miR858. SmMYB78 and AtMYB33 are targets of miR159 and miR319. AtMYB97 and AtMYB120 are targets of miR159 and miR858. SmMYB80 and AtMYB65 and AtMYB104 are regulated by miR159 and miR319 and miR858. All SmMYBs in S18 were predominantly expressed in flowers (Additional file 3: Figure S2) and AtMYBs in the subgroup were involved in anther development [33–41]. Taken together, the results suggest the conserved regulatory mechanism of miRNA-MYB module in flower development in S. miltiorrhiza and Arabidopsis.
The 38 miRNA-targeted SmMYBs distribute in 21 subgroups (Figures 1 and 5). Targets of miR159 are included in S18 and S36. Targets of miR828 belong to S6 and S15. MiR858-regulated SmMYBs widely distribute in 19 subgroups. It suggests the significance of miRNAs in regulating multiple cellular processes. Except S15 and S18, each of other 19 subgroups was regulated by only a miRNA family. It is consistent with the fact that MYBs in a subgroup show high sequence similarity and usually play redundant roles.
SmMYBs potentially involved in the biosynthesis of bioactive compounds
S. miltiorrhiza Bunge is a widely used Chinese medicinal material. The main bioactive compounds in S. miltiorrhiza are terpenoid tanshinones and phenolic acids. Various R2R3-MYBs were found to regulate the biosynthesis of terpenoids. For instance, AtMYB62 included in S20 is involved in gibberellin metabolism and signaling and affect root architecture, Pi uptake and acid phosphatase activity [66]. Grapevine VvMYB5b belonging to S5 is involved in flavonoid and terpenoid metabolism. Overexpression of VvMYB5b resulted in the down-regulation of phenylpropanoid metabolism whereas up-regulation of carotenoid metabolism [9]. Loblolly pine PtMYB14 belonging to S4 is a putative regulator of an isoprenoid-oriented response that leads to the accumulation of sesquiterpene in conifers [8]. It implies that SmMYBs belonging to subgroups 4, 5 and 20 are potential regulators of terpenoid biosynthesis in S. miltiorrhiza. Since terpenoid biosynthesis-related plant MYBs have not been systematically investigated, it is possible that some SmMYBs in other subgroups are also involved in terpenoid biosynthesis.
Phenolic acids are the other main group of bioactive compounds in S. miltiorrhiza. The main pathways leading to the formation of these compounds start with the aromatic amino acids L-phenylalanine and L-tyrosine. So far, a large number of R2R3-MYBs belonging to various subgroups, including S3, S4, S5, S6, S7, S13 and S21, have been found to affect the biosynthesis of these compounds. S3 MYBs, such as AtMYB58 and AtMYB63, were shown to activate PAL, C4H, 4CL and various other genes involved in lignin biosynthesis [43]. SmMYB37 belongs to the same subgroup and is predominately expressed in S. miltiorrhiza stems (Figure 1 and Additional file 3: Figure S2), implying the role of SmMYB37 in lignin biosynthesis. The other MYBs involved in lignin deposition are members of S13 and S21 [67, 68]. S4 includes 6 SmMYBs and 6 AtMYBs (Figure 1). AtMYB4 is a repressor of C4H. Down-regulation of AtMYB32 increased transcript levels of COMT gene involved in lignin biosynthesis and reduced the levels of DFR and ANS genes associated with proanthocyanidin biosynthesis [69]. S5 MYBs, AtMYB5 and AtMYB123, are involved in the biosynthesis of proanthocyanidins and tannins in Arabidopsis seeds. Additionally, AtMYB5 is also involved in trichome development [61, 70]. It indicate the role of 6 SmMYBs (SmMYB3, SmMYB28, SmMYB54, SmMYB76, SmMYB93 and SmMYB109) belonging to S4 and SmMYB6 included in S5 in proanthocyanidin biosynthesis. All of S6 AtMYBs, including AtMYB75, AtMYB90, AtMYB113 and AtMYB114, regulate anthocyanin biosynthesis [28], implying that SmMYB36 belonging to the same subgroup is also involved in anthocyanin biosynthesis. SmMYB97 is clustered into S7 with AtMYB11, AtMYB12 and AtMYB111 controlling flavonoid biosynthesis in Arabidopsis[24, 71]. Thus, SmMYB97 is a potential regulator of flavonol biosynthesis in S. miltiorrhiza. Further analysis of SmMYBs through genetic transformation will definitely shed lights on the regulatory mechanism of bioactive compound biosynthesis.
Conclusions
The MYB gene family is the largest transcription factor family in plants. It plays significant roles in plant development, secondary metabolism, and stress responses. The identification and characterization of 110 SmMYB genes provides a foundation for understanding the regulatory mechanism of MYBs in S. miltiorrhiza. Through the comprehensive classification and comparative analysis of the R2R3-MYB gene family in S. miltiorrhiza and Arabidopsis, we showed the conservation and diversity of MYBs in plants. The distribution of residues in the MYB domain of S. miltiorrhiza R2R3-MYBs is quite similar to Arabidopsis, although the patterns at various positions are different. A total of 40 motifs were identified in downstream of the MYB domain of SmMYBs and AtMYBs. Among them, 35 exist in both SmMYBs and AtMYBs, 5 are AtMYB-specific. Based on the phylogenetic tree and previous results from Arabidopsis[10], SmMYBs and AtMYBs were classified into 37 subgroups, of which 31 include proteins from S. miltiorrhiza and Arabidopsis, whereas 2 are specific to S. miltiorrhiza and 4 are specific to Arabidopsis. Many S. miltiorrhiza R2R3-MYBs in a subgroup share at least a motif. SmMYBs were differentially expressed in roots, stems, leaves and flowers of S. miltiorrhiza. The expression profiles are largely consistent with known functions of their Arabidopsis counterparts. We identified 38 SmMYBs to be targets of miRNAs belonging to the MIR159/R319, MIR828 and MIR858 families. Among them, 36 are regulated by a miRNA family, while the other 2 are co-regulated by miRNAs from two families. The regulatory mechanism of miRNA-MYB module is also conserved in Arabidopsis. MiRNA-targeted SmMYBs are widely distributed in different subgroups, suggesting the significance of miRNAs in regulating multiple cellular processes. Based on functional conservation of SmMYBs and AtMYBs, we predicted some functions of various SmMYBs. SmMYBs in S4, S5 and S20 are potential regulators of terpenoid biosynthesis, whereas those in S3, S4, S5, S6, S7, S13 and S21 appear to be involved in phenolic acid biosynthesis. Our results suggest the significant and complexity of MYB regulatory networks in S. miltiorrhiza and provide useful information for improving the growth and defense abilities and the production of bioactive compounds in S. miltiorrhiza.
Methods
Genome-wide survey of S. miltiorrhiza R2R3-MYBgenes
The protein sequences of 125 A. thaliana R2R3-MYBs described by Stracke et al. (2001) were downloaded from the Arabidopsis Information Resource (TAIR, http://www.Arabidopsis.org/) (Additional file 4: Table S2) [12]. Using the tBLASTn algorithm [72], S. miltiorrhiza genomic sequences putatively encoding R2R3-MYB genes were identified by BLAST analysis of A. thaliana R2R3-MYBs against the current assembly of the S. miltiorrhiza genome that was estimated to represent ~92% of the entire genome and ~96% of the protein-coding genes (Chen et al., unpublished). An e-value cut-off of 1e–10 was applied to the homologue recognition. Gene models were predicted as described previously [7]. First, all retrieved genomic sequences of the S. miltiorrhiza genome were used for gene prediction on the Genscan web server ((http://genes.mit.edu/GENSCAN.html). Second, the predicted gene models were BLAST-analyzed against the non-redundant protein sequence (nr) database (http://www.ncbi.nlm.nih.gov/BLAST) using the BLASTx algorithm with default parameters. Finally, the gene models were manually corrected after carefully checked the alignment between SmMYB genes and MYBs from other plant species. The MYB domain of each SmMYB protein was predicted using Pfam 26.0 on the Pfam sever with default parameters (http://pfam.sanger.ac.uk). Protein sequences with two repeats in the MYB domain were recognized as members of the R2R3-MYB subfamily.
RNA extraction, coding sequence (CDS) cloning and quantitative real-time reverse transcription-PCR (qRT-PCR)
Roots, stems, leaves and flowers of whole genome-decoded S. miltiorrhiza Bunge (line 993) were collected and stored in liquid nitrogen until use. Total RNA was extracted using the plant total RNA extraction kit (Aidlab, China). Genomic DNA contamination was eliminated by pre-treating total RNA with RNase-Free DNase (Promega, USA). RNA integrity was analyzed on a 1.2% agarose gel. RNA quantity was determined using a NanoDrop 2000C Spectrophotometer (Thermo Scientific, USA). Total RNA was reverse-transcribed by Superscript III Reverse Transcriptase (Invitrogen, USA). The full-length CDSs of 110 SmMYBs were amplified by PCR using the primers listed in Additional file 5: Table S3. PCR products were gel-purified, cloned, and then sequenced. qRT-PCRs were performed as previously described by Ma et al. using gene-specific primers (Additional file 6: Table S4) [7]. The length of amplicons was between 80 bp and 250 bp. SmUBQ10 was used as a reference gene. The data was analyzed as described previously [7].
Analysis of the MYB domain and other motifs
To analyze the features of MYB domains of S. miltiorrhiza R2R3-MYB proteins, the amino acid sequence of R2 and R3 repeats in 110 S. miltiorrhiza R2R3-MYB proteins were aligned with the ClustalW method using BioEdit software (http://www.mbio.ncsu.edu/BioEdit/bioedit.html) and adjusted manually. The sequence logos for R2 and R3 MYB repeats were created by submitting the multiple alignment sequences to the WebLogo server (http://weblogo.berkeley.edu/logo.cgi) [73]. Potential protein motifs outside the MYB domain were predicted using the MEME Suite version 3.5.7 [74]. The following parameter settings were applied. It includes the distribution of motifs: zero and one per sequence; maximum number of motifs to find: 40, minimum width of motif: 6, and maximum width of motif: 120. The motif must be present in all members within the same subgroup and only those with an e-value less than 1e–10 were kept for further analysis.
Phylogenetic tree construction
To generate the phylogenetic trees of R2R3-MYB transcription factor genes, a neighbor-joining (NJ) tree was constructed for the full-length protein sequences of 110 SmMYBs and 125 AtMYBs using MEGA version 4.0. Bootstrapping (1000 replicates) was performed to support the statistical reliability.
Identification of SmMYBswith perfect or near-perfect complementary sequences to miRNAs
Plant miRNA sequences were downloaded from miRBase (http://www.mirbase.org/) [60]. The complementary sequences of SmMYBs to miRNAs were searched using psRNATarget [64]. The maximum expectations of 3.0 and the target accessibility-allowed maximum energy to unpair the target site of 50 were applied.
Availability of supporting data
SmMYB sequences supporting the results of this article are available in GenBank under accession numbers KF059355–KF059464. Phylogenetic data supporting the results of this article are available in the TreeBASE repository, http://purl.org/phylo/treebase/phylows/study/TB2:S15578.
Abbreviations
- 4CL:
-
4-coumarate coenzyme A: ligase
- C4H:
-
Cinnamate 4-hydroxylase
- CDS:
-
Sequence coding for amino acids in protein
- MEP:
-
2-C-methyl-D-erythritol 4-phosphate
- miRNA:
-
microRNA
- MVA:
-
Mevalonate
- PAL:
-
Phenylalanine ammonia-lyase
- qRT-PCR:
-
Quantitative real-time reverse transcription-PCR
- TCM:
-
Traditional Chinese medicine.
References
Wagstaff SJ, Olmstead RG, Cantino PD: Parsimony analysis of cpDNA restriction site variation in subfamily Nepetoideae (Labiatae). Am J Bot. 1995, 82 (7): 886-892. 10.2307/2445975.
Walker JB, Sytsma KJ: Staminal evolution in the genus Salvia (Lamiaceae): Molecular phylogenetic evidence for multiple origins of the staminal lever. Ann Bot. 2007, 100 (2): 375-391. 10.1093/aob/mcl176.
Cheng TO: Danshen: a popular Chinese cardiac herbal drug. J Am Coll Cardiol. 2006, 47 (7): 1498-
Cheng TO: Cardiovascular effects of Danshen. Int J Cardiol. 2006, 121 (1): 9-22.
Li YG, Song L, Liu M, Hu ZB, Wang ZT: Advancement in analysis of Salviae miltiorrhizae Radix et Rhizoma (Danshen). J Chromatogr A. 2009, 1216 (11): 1941-1953. 10.1016/j.chroma.2008.12.032.
Wang XH, Morris-Natschke SL, Lee KH: Developments in the chemistry and biology of the bioactive constituents of Tanshen. Med Res Rev. 2007, 27 (1): 133-148. 10.1002/med.20077.
Ma Y, Yuan L, Wu B, Li X, Chen S, Lu S: Genome-wide identification and characterization of novel genes involved in terpenoid biosynthesis in Salvia miltiorrhiza. J Exp Bot. 2012, 63 (7): 2809-2823. 10.1093/jxb/err466.
Bedon F, Bomal C, Caron S, Levasseur C, Boyle B, Mansfield SD, Schmidt A, Gershenzon J, Grima-Pettenati J, Séguin A, MacKay J: Subgroup 4 R2R3-MYBs in conifer trees: gene family expansion and contribution to the isoprenoid- and flavonoid-oriented responses. J Exp Bot. 2010, 61 (14): 3847-3864. 10.1093/jxb/erq196.
Mahjoub A, Hernould M, Joubès J, Decendit A, Mars M, Barrieu F, Hamdi S, Delrot S: Overexpression of a grapevine R2R3-MYB factor in tomato affects vegetative development flower morphology and flavonoid and terpenoid metabolism. Plant Physiol Biochem. 2009, 47 (7): 551-561. 10.1016/j.plaphy.2009.02.015.
Dubos C, Stracke R, Grotewold E, Weisshaar B, Martin C, Lepiniec L: MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15 (10): 573-581. 10.1016/j.tplants.2010.06.005.
Weston K: Myb proteins in life death and differentiation. Curr Opin Genet Dev. 1998, 8 (1): 76-81. 10.1016/S0959-437X(98)80065-8.
Stracke R, Werber M, Weisshaar B: The R2R3-MYB gene family in Arabidopsis thaliana. Curr Opin Plant Biol. 2001, 4 (5): 447-456. 10.1016/S1369-5266(00)00199-0.
Feller A, Machemer K, Braun EL, Grotewold E: Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J. 2011, 66 (1): 94-116. 10.1111/j.1365-313X.2010.04459.x.
Rosinski JA, Atchley WR: Molecular evolution of the Myb family of transcription factors: evidence for polyphyletic origin. J Mol Evo. 1998, 46 (1): 74-83. 10.1007/PL00006285.
Wilkins O, Nahal H, Foong J, Provart NJ, Campbell MM: Expansion and diversification of the Populus R2R3-MYB family of transcription factors. Plant Physiol. 2009, 149 (2): 981-993.
Du H, Yang SS, Liang Z, Feng BR, Liu L, Huang YB, Tang YX: Genome-wide analysis of the MYB transcription factor superfamily in soybean. BMC Plant Biol. 2012, 12 (1): 106-10.1186/1471-2229-12-106.
Du H, Feng BR, Yang SS, Huang YB, Tang YX: The R2R3-MYB transcription factor gene family in maize. PLoS One. 2012, 7 (6): e37463-10.1371/journal.pone.0037463.
Dai X, Wang Y, Yang A, Zhang WH: OsMYB2P-1 an R2R3 MYB transcription factor is involved in the regulation of phosphate-starvation responses and root architecture in rice. Plant Physiol. 2012, 159 (1): 169-183. 10.1104/pp.112.194217.
Fornalé S, Shi X, Chai C, Encina A, Irar S, Capellades M, Fuguet E, Torres JL, Rovira P, Puigdomènech P, Rigau J, Grotewold E, Gray J, Caparrós-Ruiz D: ZmMYB31 directly represses maize lignin genes and redirects the phenylpropanoid metabolic flux. Plant J. 2010, 64 (4): 633-644. 10.1111/j.1365-313X.2010.04363.x.
Zhang L, Zhao G, Xia C, Jia J, Liu X, Kong X: A wheat R2R3-MYB gene TaMYB30-B improves drought stress tolerance in transgenic Arabidopsis. J Exp Bot. 2012, 63 (16): 5873-5885. 10.1093/jxb/ers237.
McCarthy RL, Zhong R, Fowler S, Lyskowski D, Piyasena H, Carleton K, Spicer C, Ye ZH: The poplar MYB transcription factors PtrMYB3 and PtrMYB20 are involved in the regulation of secondary wall biosynthesis. Plant Cell Physiol. 2010, 51 (6): 1084-1090. 10.1093/pcp/pcq064.
Legay S, Sivadon P, Blervacq AS, Pavy N, Baghdady A, Tremblay L, Levasseur C, Ladouce N, Lapierre C, Séguin A, Hawkins S, Mackay J, Grima-Pettenati J: EgMYB1 an R2R3 MYB transcription factor from eucalyptus negatively regulates secondary cell wall formation in Arabidopsis and poplar. New Phytol. 2010, 188 (3): 774-786. 10.1111/j.1469-8137.2010.03432.x.
Nakatsuka T, Haruta KS, Pitaksutheepong C, Abe Y, Kakizaki Y, Yamamoto K, Shimada N, Yamamura S, Nishihara M: Identification and characterization of R2R3-MYB and bHLH transcription factors regulating anthocyanin biosynthesis in gentian flowers. Plant Cell Physiol. 2008, 49 (12): 1818-1829. 10.1093/pcp/pcn163.
Stracke R, Ishihara H, Huep G, Barsch A, Mehrtens F, Niehaus K, Weisshaar B: Differential regulation of closely related R2R3-MYB transcription factors controls flavonol accumulation in different parts of the Arabidopsis thaliana seedling. Plant J. 2007, 50 (4): 660-677. 10.1111/j.1365-313X.2007.03078.x.
Czemmel S, Stracke R, Weisshaar B, Cordon N, Harris NN, Walker AR, Robinson SP, Bogs J: The grapevine R2R3-MYB transcription factor VvMYBF1 regulates flavonol synthesis in developing grape berries. Plant Physiol. 2009, 151 (3): 1513-1530. 10.1104/pp.109.142059.
Ballester AR, Molthoff J, De Vos R, Hekkert BL, Orzaez D, Fernández-Moreno JP, Tripodi P, Grandillo S, Martin C, Heldens J, Ykema M, Granell A, Bovy A: Biochemical and molecular analysis of pink tomatoes: deregulated expression of the gene encoding transcription factor SlMYB12 leads to pink tomato fruit color. Plant Physiol. 2010, 152 (1): 71-84. 10.1104/pp.109.147322.
Nakatsuka T, Saito M, Yamada E, Fujita K, Kakizaki Y, Nishihara M: Isolation and characterization of GtMYBP3 and GtMYBP4 orthologues of R2R3-MYB transcription factors that regulate early flavonoid biosynthesis in gentian flowers. J Exp Bot. 2012, 63 (18): 6505-6517. 10.1093/jxb/ers306.
Gonzalez A, Zhao M, Leavitt JM, Lloyd AM: Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. Plant J. 2008, 53 (5): 814-827. 10.1111/j.1365-313X.2007.03373.x.
Chiu LW, Li L: Characterization of the regulatory network of BoMYB2 in controlling anthocyanin biosynthesis in purple cauliflower. Planta. 2012, 236 (4): 1153-1164. 10.1007/s00425-012-1665-3.
Chagne D, Lin-Wang K, Espley RV, Volz RK, How NM, Rouse S, Brendolise C, Carlisle CM, Kumar S, Desilva N, Micheletti D, McGhie T, Crowhurst RN, Storey RD, Velasco R, Hellens RP, Gardiner SE, Allan AC: An ancient duplication of apple MYB transcription factors is responsible for novel red fruit-flesh phenotypes. Plant Physiol. 2013, 161 (1): 225-239. 10.1104/pp.112.206771.
Feng S, Wang Y, Yang S, Xu Y, Chen X: Anthocyanin biosynthesis in pears is regulated by a R2R3-MYB transcription factor PyMYB10. Planta. 2010, 232 (1): 245-255. 10.1007/s00425-010-1170-5.
Zhang L, Zhao G, Jia J, Liu X, Kong X: Molecular characterization of 60 isolated wheat MYB genes and analysis of their expression during abiotic stress. J Exp Bot. 2012, 63 (1): 203-214. 10.1093/jxb/err264.
Gubler F, Kalla R, Roberts JK, Jacobsen JV: Gibberellin-regulated expression of a myb gene in barley aleurone cells: evidence for Myb transactivation of a high pI alpha-amylase gene promoter. Plant Cell. 1995, 7 (11): 1879-1891.
Gocal GF, Poole AT, Gubler F, Watts RJ, Blundell C, King RW: Long-day up-regulation of a GAMYB gene during Lolium temulentum inflorescence formation. Plant Physiol. 1999, 119 (4): 1271-1278. 10.1104/pp.119.4.1271.
Tsuji H, Aya K, Ueguchi-Tanaka M, Shimada Y, Nakazono M, Watanabe R, Nishizawa NK, Gomi K, Shimada A, Kitano H, Ashikari M, Matsuoka M: GAMYB controls different sets of genes and is differentially regulated by microRNA in aleurone cells and anthers. Plant J. 2006, 47 (3): 427-444. 10.1111/j.1365-313X.2006.02795.x.
Aya K, Ueguchi-Tanaka M, Kondo M, Hamada K, Yano K, Nishimura M, Matsuoka M: Gibberellin modulates anther development in rice via the transcriptional regulation of GAMYB. Plant Cell. 2009, 21 (5): 1453-1472. 10.1105/tpc.108.062935.
Gocal GF, Sheldon CC, Gubler F, Moritz T, Bagnall DJ, MacMillan CP, Li SF, Parish RW, Dennis ES, Weigel D, King RW: GAMYB–like genes flowering and gibberellin signaling in Arabidopsis. Plant Physiol. 2001, 127 (4): 1682-1693. 10.1104/pp.010442.
Murray F, Kalla R, Jacobsen J, Gubler F: A role for HvGAMYB in anther development. Plant J. 2003, 33 (3): 481-491. 10.1046/j.1365-313X.2003.01641.x.
Kaneko M, Inukai Y, Ueguchi-Tanaka M, Itoh H, Izawa T, Kobayashi Y, Hattori T, Miyao A, Hirochika H, Ashikari M, Matsuoka M: Loss-of-function mutations of the rice GAMYB gene impair α-amylase expression in aleurone and flower development. Plant Cell. 2004, 16 (1): 33-44. 10.1105/tpc.017327.
Gong X, Bewley DJ: A GAMYB-like gene in tomato and its expression during seed germination. Planta. 2008, 228 (4): 563-572. 10.1007/s00425-008-0759-4.
Millar AA, Gubler F: The Arabidopsis GAMYB-like genes MYB33 and MYB65 are microRNA-regulated genes that redundantly facilitate anther development. Plant Cell. 2005, 17 (3): 705-721. 10.1105/tpc.104.027920.
Alonso-Peral MM, Li J, Li Y, Allen RS, Schnippenkoetter W, Ohms S, White RG, Millar AA: The microRNA159-regulated GAMYB-like genes inhibit growth and promote programmed cell death in Arabidopsis. Plant Physiol. 2010, 154 (2): 757-771. 10.1104/pp.110.160630.
Zhou J, Lee C, Zhong R, Ye ZH: MYB58 and MYB63 are transcriptional activators of the lignin biosynthetic pathway during secondary cell wall formation in Arabidopsis. Plant Cell. 2009, 21 (1): 248-266. 10.1105/tpc.108.063321.
Müller D, Schmitz G, Theres K: Blind homologous R2R3 Myb genes control the pattern of lateral meristem initiation in Arabidopsis. Plant Cell. 2006, 18 (3): 586-597. 10.1105/tpc.105.038745.
Keller T, Abbott J, Moritz T, Doerner P: Arabidopsis REGULATOR OF AXILLARY MERISTEMS1 controls a leaf axil stem cell niche and modulates vegetative development. Plant Cell. 2006, 18 (3): 598-611. 10.1105/tpc.105.038588.
Feng C, Andreasson E, Maslak A, Mock HP, Mattsson O, Mundy J: Arabidopsis MYB68 in development and responses to environmental cues. Plant Sci. 2004, 167 (5): 1099-1107. 10.1016/j.plantsci.2004.06.014.
Noda K, Glover BJ, Linstead P, Martin C: Flower colour intensity depends on specialized cell shape controlled by a Myb-related transcription factor. Nature. 1994, 369 (6482): 661-664. 10.1038/369661a0.
Perez-Rodriguez M, Jaffe FW, Butelli E, Glover BJ, Martin C: Development of three different cell types is associated with the activity of a specific MYB transcription factor in the ventral petal of Antirrhinum majus flowers. Development. 2005, 132 (2): 359-370. 10.1242/dev.01584.
Baumann K, Perez–Rodriguez M, Bradley D, Venail J, Bailey P, Jin H, Koes R, Roberts K, Martin C: Control of cell and petal morphogenesis by R2R3 MYB transcription factors. Development. 2007, 134 (9): 1691-1701. 10.1242/dev.02836.
Jaffé FW, Tattersall A, Glover BJ: A truncated MYB transcription factor from Antirrhinum majus regulates epidermal cell outgrowth. J Exp Bot. 2007, 58 (6): 1515-1524. 10.1093/jxb/erm020.
Jakoby MJ, Falkenhan D, Mader MT, Brininstool G, Wischnitzki E, Platz N, Hudson A, Hülskamp M, Larkin J, Schnittger A: Transcriptional profiling of mature Arabidopsis trichomes reveals that NOECK encodes the MIXTA-like transcriptional regulator MYB106. Plant Physiol. 2008, 148 (3): 1583-1602. 10.1104/pp.108.126979.
Woodger FJ, Gubler F, Pogson BJ, Jacobsen JV: A Mak-like kinase is a repressor of GAMYB in barley aleurone. Plant J. 2003, 33 (4): 707-717. 10.1046/j.1365-313X.2003.01663.x.
Jones-Rhoades MW, Bartel DP, Bartel B: MicroRNAs and their regulatory roles in plants. Annu Rev Plant Biol. 2006, 57: 19-53. 10.1146/annurev.arplant.57.032905.105218.
Chen X: Small RNAs and their roles in plant development. Annu Rev Cell Dev Biol. 2009, 25: 21-44. 10.1146/annurev.cellbio.042308.113417.
Voinnet O: Origin biogenesis and activity of plant microRNAs. Cell. 2009, 136 (4): 669-687. 10.1016/j.cell.2009.01.046.
Lu S, Sun YH, Chiang VL: Stress-responsive microRNAs in Populus. Plant J. 2008, 55 (1): 131-151. 10.1111/j.1365-313X.2008.03497.x.
Lu S, Sun YH, Shi R, Clark C, Li L, Chiang VL: Novel and mechanical stress responsive microRNAs in Populus trichocarpa that are absent from Arabidopsis. Plant Cell. 2005, 17 (8): 2186-2203. 10.1105/tpc.105.033456.
Lu S, Yang C, Chiang VL: Conservation and diversity of microRNA associated copper-regulatory networks in Populus trichocarpa. J Integr Plant Biol. 2011, 53 (11): 879-891. 10.1111/j.1744-7909.2011.01080.x.
Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B, Bartel DP: Prediction of plant microRNA targets. Cell. 2002, 110 (4): 513-520. 10.1016/S0092-8674(02)00863-2.
Kozomara A, Griffiths-Jones S: miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39 (Database Issue): D152-D157.
Rajagopalan R, Vaucheret H, Trejo J, Bartel D: A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 2006, 20 (24): 3407-3425. 10.1101/gad.1476406.
Allen RS, Li J, Stahle MI, Dubroué A, Gubler F, Millar AA: Genetic analysis reveals functional redundancy and the major target genes of the Arabidopsis miR159 family. Proc Natl Acad Sci USA. 2007, 104 (41): 16371-16376. 10.1073/pnas.0707653104.
Xia R, Zhu H, An YQ, Beers EP, Liu Z: Apple miRNAs and tasiRNAs with novel regulatory networks. Genome Biol. 2012, 13 (6): R47-10.1186/gb-2012-13-6-r47.
Dai X, Zhao PX: psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 2011, 39 (Web Server issue): W155-W159.
Tominaga-Wada R, Nukumizu Y, Sato S, Kato T, Tabata S, Wada T: Functional divergence of MYB-related genes WEREWOLF and AtMYB23 in Arabidopsis. Biosci Biotechnol Biochem. 2012, 76 (5): 883-887. 10.1271/bbb.110811.
Devaiah BN, Madhuvanthi R, Karthikeyan AS, Raghothama KG: Phosphate starvation responses and gibberellic acid biosynthesis are regulated by the MYB62 transcription factor in Arabidopsis. Mol Plant. 2009, 2 (1): 43-58. 10.1093/mp/ssn081.
Newman LJ, Perazza DE, Juda L, Campbell MM: Involvement of the R2R3-MYB AtMYB61 in the ectopic lignification and dark-photomorphogenic components of the det3 mutant phenotype. Plant J. 2004, 37 (2): 239-250. 10.1046/j.1365-313X.2003.01953.x.
Zhong RZ, Lee C, Zhou J, McCarthy RL, Ye ZH: A battery of transcription factors involved in the regulation of secondary cell wall biosynthesis in Arabidopsis. Plant Cell. 2008, 20 (10): 2763-2782. 10.1105/tpc.108.061325.
Preston J, Wheeler J, Heazlewood J, Li SF, Parish RW: AtMYB32 is required for normal pollen development in Arabidopsis thaliana. Plant J. 2004, 40 (6): 979-995. 10.1111/j.1365-313X.2004.02280.x.
Gonzalez A, Mendenhall J, Huo Y, Lloyd A: TTG1 complex MYBs MYB5 and TT2 control outer seed coat differentiation. Dev Biol. 2009, 325 (2): 412-421. 10.1016/j.ydbio.2008.10.005.
Mehrtens F, Kranz H, Bednarek P, Weissharr B: The Arabidopsis transcription factor MYB12 is a flavonol-specific regulator of phenylpropanoid biosynthesis. Plant Physiol. 2005, 138 (2): 1083-1096. 10.1104/pp.104.058032.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25 (17): 3389-3402. 10.1093/nar/25.17.3389.
Crooks GE, Hon G, Chandonia JM, Brenner SE: WebLogo: A sequence logo generator. Genome Res. 2004, 14 (6): 1188-1190. 10.1101/gr.849004.
Bailey TL, Bodén M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS: MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009, 37 (Web Server issue): W202-W208.
Acknowledgements
We thank Prof. Shilin Chen and the sequencing group in our institute for kindly providing the S. miltiorrhiza genome sequence. We appreciate Prof. Chang Liu for aiding in sequence analysis and Prof. Xian’en Li for providing S. miltiorrhiza plants. This work was supported by grants from the Natural Science Foundation of China (grant no. 31370327), the Beijing Natural Science Foundation (grant no. 5112026), the Major Scientific and Technological Special Project for Significant New Drugs Creation (grant no. 2012ZX09301002-001-031), the Program for Changjiang Scholars and Innovative Research Team in University (grant no. IRT1150), the Research Fund for the Doctoral Program of Higher Education of China (grant no. 20111106110033), and the Program for Xiehe Scholars in Chinese Academy of Medical Sciences & Peking Union Medical College.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CL contributed to RNA extraction, coding sequence (CDS) cloning, qRT-PCR and bioinformatics analysis, and participated in writing the manuscript. SL designed the experiment, participant in bioinformatics analysis, and wrote the manuscript. Both authors have read and approved the version of manuscript.
Electronic supplementary material
12864_2013_5925_MOESM1_ESM.xls
Additional file 1: Table S1: Sequence features of R2R3-MYBs in S. miltiorrhiza. Some sequence features of R2R3-MYBs in S. miltiorrhiza are shown. (XLS 153 KB)
12864_2013_5925_MOESM2_ESM.doc
Additional file 2: Figure S1: Architecture of conserved protein motifs in SmMYBs and AtMYBs. Conserved motifs are indicated in numbered color boxes. (DOC 5 MB)
12864_2013_5925_MOESM3_ESM.doc
Additional file 3: Figure S2: Expression patterns of SmMYB genes in various tissues of S. miltiorrhiza. Fold changes of transcript levels in root (RT), stems (St), leaves (Le) and flowers (Fl) of S. miltiorrhiza plants are shown. SmMYBs expression is relative to SmUBQ10*10000. (DOC 1 MB)
12864_2013_5925_MOESM4_ESM.xls
Additional file 4: Table S2: Sequence features of R2R3-MYBs in A. thaliana. Some sequence features of R2R3-MYBs in A. thaliana are shown. (XLS 82 KB)
12864_2013_5925_MOESM5_ESM.xls
Additional file 5: Table S3: Primers used for cloning of SmMYB CDSs. Complete set of primers used for amplification of SmMYB CDSs. (XLS 58 KB)
12864_2013_5925_MOESM6_ESM.xls
Additional file 6: Table S4: Primers used for qRT-PCR analysis of SmMYB genes. Complete set of primers used for qRT-PCR. (XLS 45 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited.
About this article
Cite this article
Li, C., Lu, S. Genome-wide characterization and comparative analysis of R2R3-MYB transcription factors shows the complexity of MYB-associated regulatory networks in Salvia miltiorrhiza. BMC Genomics 15, 277 (2014). https://doi.org/10.1186/1471-2164-15-277
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-277