
Abstract
Background
Genetic variations in TGFB1 gene have been studied in relation to coronary heart disease (CHD) risk, but the results were inconsistent.
Methods
We performed a systematic review of published studies on the potential role of TGFB1 genetic variation in CHD risk. Articles that reported the association of TGFB1 genetic variants with CHD as primary outcome were searched via Medline and HuGE Navigator through July 2011. The reference lists from included articles were also reviewed.
Results
Data were available from 4 studies involving 1777 cases and 7172 controls for rs1800468, 7 studies involving 5935 cases and 10677 controls for rs1800469, 7 studies involving 6634 cases and 9620 controls for rs1982073, 5 studies involving 5452 cases and 9999 controls for rs1800471, and 4 studies involving 5143 cases and 4229 controls for rs1800472. The pooled odds ratios (ORs) for CHD among minor T allele carriers of rs1800469, minor C allele carriers of rs1982073, and minor C allele carriers of rs1800471 versus homozygous major allele carriers was 1.14 (95% confidence interval [CI]: 1.05-1.24), 1.18 (95% CI: 1.04-1.35), and 1.16 (95% CI: 1.02-1.32), respectively. No substantial heterogeneity for ORs was detected among the included Caucasian populations for all SNPs. However, for rs1800471, the statistical significance disappeared after adjusting for potential publication bias. No significant association was found between rs1800468 and rs1800472 variants and CHD risk.
Conclusion
Minor allele carriers of two genetic variants (rs1800469 and rs1982073) in TGFB1 have a 15% increased risk of CHD.
Similar content being viewed by others

Background
Transforming growth factor-β1 (TGFβ1) is a ubiquitously expressed multifunctional cytokine that is involved in many physiological and pathological processes. TGFβ1 has been demonstrated to be of fundamental importance in the development, physiology and pathology of the vascular system. Research into the mechanisms of TGFβ1 signaling over the past two decades has led to the development of a well-accepted canonical signaling cascade involving heterotetrameric complexes of type I and type II serine/threonine-kinase transmembrane receptors together with Smad transcription factors that act as intracellular signaling effectors. However, the exact mechanisms by which TGFβ1 signaling exerts its effects within the vasculature are still incompletely understood [1–4]. According to the literature [2–7], TGFβ1 can be secreted by several cell types, including peripheral blood mononuclear cells, macrophages, platelets, endothelial cells, vascular smooth muscle cells (VSMCs), myofibroblasts, and renal cells. Its regulatory function on the vessel wall is directed at endothelial cells, VSMC and extracellular matrix [1–3, 5–8]. Although the role of TGFβ1 in the pathogenesis of atherosclerosis is being recognized, the association between plasma TGFβ1 levels and coronary heart disease (CHD) risk is still controversial [6, 9–11]. There may be several explanations for the controversy: 1), TGFβ1 is a bimodal regulator of both endothelial cells and VSMC proliferation, depending on local TGFβ1 levels, cell density, and/or membrane TGFβ receptors [1, 2, 5, 7, 8, 12]; 2), different pathophysiological stages of CHD may differentially affect the biological effects of TGFβ1 [1, 3, 10]; and 3), circulating TGFβ1 levels may not reflect the real vascular interstitial TGFβ1 levels that are directly involved in the pathogenesis of CHD [3–5, 13]. Also, animal-model studies of CHD reported inconsistent findings on the role of TGFβ1 in CHD development. This might, however, be due to the dysregulated systemic immune function from different methods used, i.e. injecting TGFβ1 antibodies, infusing a soluble TGFβ receptor, or using transgenic or knockout mice [6, 8].
Although the amino acid sequence of the active form of TGFβ1 is highly conserved across mammalian species [7, 14, 15], common TGFB1 genetic variations that could cause variable constitutive or induced expression of TGFB1 or protein structural changes and, as a result, changed TGFβ1 activity, have been identified. They include rs1800468 (−800 G/A) and rs1800469 (−509 C/T) in the promoter region, rs1982073 (868 T/C, Leu10Pro) and rs1800471 (913 G/C, Arg25Pro) in the signal peptide region, and rs1800472 (11929 C/T, Thr263Ile) in the region encoding the precursor part of the protein [4, 15–19]. These genetic variants are generally in strong linkage disequilibrium (LD) with each other, and this DNA LD block covers the whole 5′ proximal region of the TGFB1 gene in Caucasian populations [4, 14, 16, 17]. The minor alleles of these genetic variants or the haplotypes where the minor alleles are located, were associated with increased CHD risk in some [11, 16, 20], but not all studies [10, 14, 21, 22], and even an opposite association has been observed [23]. This may partly be explained by a relatively small sample size, different CHD endpoints and/or different study populations in each of the published studies. Demonstrating an association may require a much larger number of subjects, which may be beyond the resource of one single study. Multiple replicated loci have recently been identified from genome-wide association (GWA) studies of CHD. However, they together explain only a small part of its heritability [24, 25]. It has been suggested that the adopted highly stringent statistical criteria and/or the imperfect coverage of genetic variants by current GWA studies might prevent the discovery of potential loci associated with CHD risk [26]. No meta-analysis describing TGFB1 genetic variants in relation to CHD risk exists; therefore, we performed a meta-analysis of published data that tested for TGFB1 genetic variants associated with CHD risk.
Methods
Selection criteria
For inclusion, studies 1) had to be case–control or cohort in design, 2) examined the association between TGFB1 gene polymorphisms and primary outcomes of CHD, coronary artery disease or myocardial infarction (MI), 3) used validated coronary heart disease phenotypes (diagnostic criteria included angiographical confirmation; elevations of cardiac enzymes, changes of electrocardiographic and clinical symptoms according to the World Health Organizations criteria; a documented history of coronary artery bypass graft, percutaneous transluminal coronary angioplasty, or percutaneous coronary intervention), and 4) involved unrelated participants.
Search strategy
All studies reporting on the association between TGFB1 gene polymorphism and CHD risk published before July 2011 were identified by comprehensive electronic searches of Medline and HuGENet. Terms used for the searches were “TGFB1,” “ischemic heart disease,” “coronary heart disease,” “coronary artery disease,” “acute coronary syndrome,” “myocardial infarction,” and “angina pectoris” combined with “gene,” “genetic,” “variant,” “mutation” or “polymorphism.” Hand searches for related articles among the reference lists of included articles were also performed. If essential information of a study was not presented in the publication, authors were contacted for details. The study was excluded if the information could not be obtained. In addition, the relevant data from the two latest large-scale meta-analyses of CHD GWA studies (PROCARDIS [25] and CARDIoGRAM [24]; Additional file ( 1)) were also included for part of the analysis.
Data extraction
The first author, published year, country, study population, mean age of participants, gender distribution, study design, sample size, outcome, diagnostic criteria, genotyping method, characteristics of the controls, allele frequencies, and genotype distributions were extracted. In PROCARDIS [25] and CARDIoGRAM [24], the studied SNPs were not available in the genome-wide genotyping assay and had to be imputed. The SNPs imputed with high quality (MACH_R2 > 0.3) were included in the analyses.
Statistical analysis
Deviance from Hardy-Weinberg equilibrium (HWE) was assessed for the controls of each study using Fisher’s exact test. For an Iranian study [11], only data on rs1982073 was utilized in the meta-analysis because other SNPs deviated from HWE. Genotype distributions of controls for studies with case–control design or the entire group for studies with cohort design were used to estimate the frequency of the putative risk allele for each SNP using the inverse variance method (Additional file 2) [27]. Crude ORs with 95% confidence intervals (CI) were used to evaluate the association between genetic polymorphisms and CHD risk. Pooled ORs were calculated for several genetic models, i.e. the co-dominant model, the dominant model, and the recessive model. Since the co-dominant model effects (or additive model effects) cannot be straightforward calculated from the extracted summary data from each study, we presented the effects of two groups of genotype comparison (Additional file 2). Since the using of best-guess genotype from the genotype imputation process can lead to both false positives and loss of power [28], the allele effects from the PROCARDIS [25] and CARDIoGRAM [24] studies were only included in part of the co-dominant model analyses (Additional file 2). Statistical heterogeneity in the ORs across studies was assessed with the Q-test. If there was heterogeneity, ethnicity as source of heterogeneity was explored by pooling the data from Caucasian populations only. If there was no heterogeneity, the fixed-effect model was used to evaluate the overall gene effect; otherwise, the random-effect model was used. Presence of publication bias was explored with Begg’s funnel plot and Egger’s regression test. If potential publication bias existed, the Duval and Tweedie nonparametric “trim and fill” method [29] was used to adjust for it. All reported p values were two-tailed, and statistical significance was defined at the α = 0.05 level. All analyses were performed with the R metafor package [30].
Results
Study inclusion and characteristics
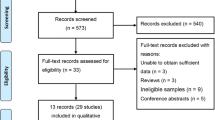
Fifteen citations were identified through the original literature search; none were meta-analyses [10, 11, 14, 16, 20–23, 31–37]. After full review, two studies were excluded because they were conducted in patient cohorts with a composite end-point that included CHD [31, 32]. Three studies (two European [10, 33] and one Japanese [34]) were conducted without “proper” controls of no CHD history and were also excluded. One additional Chinese study met the inclusion criteria but was excluded due to unavailability of essential information even after contacting the authors [35]. The 9 remaining studies, together with PROCARDIS [25] and CARDIoGRAM [24] studies, were included in the meta-analysis (Table 1) [11, 14, 16, 20–23, 36, 37].
Quantitative synthesis
None of the genetic variants were associated with CHD risk when applying a recessive model (data not shown). Rs1800468 and rs1800472 were not associated with CHD in either co-dominant or dominant model (Additional file 2).
For rs1800469, both the CT genotype in the co-dominant model and the presence of the minor T allele in the dominant model conferred a risk for CHD when compared to the common CC genotype (OR = 1.14, 95% CI: 1.04-1.25; and OR = 1.14, 95% CI: 1.05-1.24, respectively). The TT genotype conferred a non-significant risk of similar magnitude (Figure 1). For rs1982073, the TC genotype conferred a risk for CHD in the co-dominant model (OR = 1.18, 95% CI: 1.08-1.28), but the CC genotype did not when compared to the common TT genotype (Figure 2). Under a dominant model, the presence of the minor C allele was associated with a 1.18 times increased risk for CHD (Additional file 2).

Meta-analysis for coronary heart disease risk depending on the rs1800469 (−509 C/T) polymorphism in the TGFB1 gene. ORs and corresponding 95% confidence intervals (CIs) are shown. Fixed effects were reported because no significant heterogeneity between studies was observed. A. Comparison of the homozygous TT genotype with the wild type CC genotype (p = 0.08); B. Comparison of the heterozygous CT genotype with the wild type CC genotype (p = 0.004); C. Comparison of the TT + CT genotype with the wild type CC genotype (p = 0.003).

Meta-analysis for coronary heart disease risk depending on the rs1982073 (868 T/C) polymorphism in the TGFB1 gene. ORs and corresponding 95% confidence intervals (CIs) are shown. A. Comparison of the homozygous CC genotype with the wild type TT genotype (random-effect model, p = 0.26); B. Comparison of the heterozygous TC genotype with the wild type TT genotype (random-effect model, p = 0.0002); C. Comparison of the CC + TC genotype with the wild type TT genotype (random-effect model, p = 0.01).
For rs1800471, both the GC genotype in the co-dominant model and the presence of the minor C allele in the dominant model conferred a risk for CHD when compared to the common GG genotype (OR = 1.15, 95% CI: 1.01-1.31; and OR = 1.16, 95% CI: 1.02-1.32, respectively). The CC genotype conferred a 1.25 times increased risk, but this was not statistically significant (Figure 3).

Meta-analysis for coronary heart disease risk depending on the rs1800471 (913 G/C) polymorphism in the TGFB1 gene. ORs and corresponding 95% confidence intervals (CIs) are shown. Fixed effects were reported because no significant heterogeneity between studies was observed. A. Comparison of the homozygous CC genotype with the wild type GG genotype (p = 0.49); B. Comparison of the heterozygous GC genotype with the wild type GG genotype (p = 0.03); C. Comparison of the GC + CC genotype with the wild type GG genotype (p = 0.02).
After adjusting for multiple testing using Bonferroni correction, all significant associations for rs1800469 and rs1982073 under the co-dominant and dominant models remained. However, for rs1800471, associations were no longer statistically significant (p > 0.017 in Additional file 2). When the relevant allele effects from the PROCARDIS [25] and CARDIoGRAM [24] studies were included in the co-dominant model analyses, the aforementioned associations attenuated; however, the association for rs1982073 persisted (Additional file 2).
No substantial heterogeneity for the ORs was detected among the included Caucasian populations (Additional file 2). For rs1982073, however, some heterogeneity existed between Caucasian populations and non-Caucasian populations with regard to both CC and CC + TC vs. TT contrasts (Additional file 2).
Sensitivity analysis and publication bias
To evaluate the influence of the individual studies on the pooled ORs for rs1800469, rs1982073 and rs1800471, each time, a single study involved in the meta-analysis was deleted. The Rotterdam study [21] and the Japanese study [23] tended to attenuate the pooled ORs in the co-dominant model for rs1800469 and rs1982073, respectively (Additional file 3). No substantial alteration in the observed pooled ORs was observed for rs1800471 (Additional file 3). Begg’s funnel plot and Egger’s regression test were performed to assess potential publication bias for rs1800469, rs1982073 and rs1800471. Although the P values for Egger’s regression tests for all investigated models were > 0.05, Begg’s funnel plot still suggested a certain degree of publication bias, potentially from small studies with significant positive results (Additional file 4). After performing the “trim and fill method” to adjust for potential publication bias, the results for rs1800469 and rs1982073 did not change significantly (data not shown). However, for rs1800471, the statistical significance for both co-dominant and dominant models disappeared (P = 0.07).
Discussion
Several studies have been carried out to test the hypothesis that genetic polymorphisms in the TGFB1 gene including rs1800468, rs1800469, rs1982073, rs1800471 and rs1800472 might be associated with CHD risk, but data have yielded conflicting results. Possible concerns in genetic association studies are that a positive association might be spurious, while a negative result might be due to a small sample size. In this meta-analysis, we incorporated all eligible studies to date and provided some evidence that rs1800469 and rs1982073 in the TGFB1 gene are associated with CHD risk in Caucasian populations. The inconsistency between the previously reported results for these SNPs might be due to the small sample sizes in most of the studies, especially in combination with a modest effect.
Circulating TGFβ1 levels are predominantly under genetic control with a heritability of 0.54 [17]. Both the CHD-associated minor risk alleles of rs1800469 and rs1982073 correlate with an increase in gene expression, TGFβ1 secretion, and plasma TGFβ1 levels [4, 11, 15, 17, 19, 23]. These similar observations might be due to the strong LD between them [4, 14, 16, 17]. Shah et al. [18] demonstrated exclusively in vivo and in vitro recruitment of transcription regulator AP1 to -509 C (the major non-risk allele of rs1800469) leading to transcriptional repression of the TGFB1 gene. However, the exact functional variant in this gene region merits further identification. In support of the aforementioned positive association between CHD risk alleles of TGFB1 and increased TGFβ1 production, increased TGFβ1 levels were observed in different stages of plaque development in some histological studies [7, 38–40]. In addition, enhanced TGFβ1 signalling is established to cause cartilaginous metaplasia of vascular media and progressive intima-media thickening after vascular injuries [2–5, 7, 8, 12]. Interestingly, an increased TGFβ1 regulated gene expression was observed in both atherosclerotic and restenotic lesions [41]. Recently, the TGFβ1 signalling pathway is suggested to be involved in the genetic determining of CHD for the most replicated 9p21.3 locus [42–44]. A genetic variant in the SMAD3 gene that encodes one of the downstream activating transcriptional mediators (Smad3) of TGFβ1 signalling [1, 2] was associated with CHD risk in a GWA study [44], which was recently replicated in a large-scale meta-analysis of CHD studies [45].
It has previously been shown that abnormal enlargement of human coronary arteries (positive remodelling and aneurysmal coronary lesions) occurs in response to the development of intimal plaque [46–49], which is correlated with future acute coronary syndromes and cardiac events [50, 51]. A strong heritable component (h 2 = 0.52) of such abnormal enlargement of the coronary artery in the pathogenesis of coronary artery disease was observed, especially in the proximal coronary artery [52, 53]. Interestingly, the 9p21.3 locus is also associated with increased risk of abdominal aortic aneurysm [54–56] and intracranial aneurysm [57, 58]. Recently, high plasma TGFβ1 levels have been implicated in the manifestation of aortic root dilation in Marfan syndrome [6, 13, 59]. Furthermore, genetic variations along the TGFβ1 signalling pathway are associated with coronary artery aneurysm formation and aortic root dilation in Kawasaki diseases [60], whereas mutations in genes of TGFβ1 signalling pathway (TGFBR1, TGFBR2[61, 62], and SMAD3[63]) or TGFβ1 inhibitor genes [64] are implicated in familial or syndromic forms of thoracic aortic aneurysms and dissection. Taken together, this points at altered vascular remodelling from increased TGFβ1 signalling in the pathogenesis of CHD. However, given the fact that TGFβ1 is produced by multiple lineages of resident cells in vascular wall and atherosclerotic lesion and the fact that it acts in an autocrine, paracrine, and endocrine fashion [4], it has been very difficult to pinpoint the exact cellular sources of TGFβ1 that are relevant for the pathogenesis of CHD. More research on this topic is warranted.
Some limitations of this meta-analysis should be acknowledged. First, a relatively small number of studies for each SNP was included, and therefore we cannot rule out heterogeneity completely in Caucasian populations although most of the P values for Q-tests were > 0.05. Second, the results in the co-dominant model for rs1800469 and rs1982073 were dominated by the Rotterdam [21] and the Japanese study [23] as shown by the sensitivity analysis. However, this did not affect our main conclusions. Third, the potential publication bias of relatively small sample-sized studies might have affected the results, as there might be eligible studies with negative results that were not published. In the context of these limitations, our current results should be interpreted with caution.
Conclusion
In conclusion, the present study demonstrates an association between rs1800469 and rs1982073 in the TGFB1 gene and CHD risk in Caucasian populations. Enhanced TGFβ1 signalling may therefore be involved in the pathogenesis of CHD.
Abbreviations
- CHD:
-
Coronary heart disease
- CI:
-
Confidence interval
- GWA:
-
Genome-wide association
- HWE:
-
Hardy-Weinberg equilibrium
- LD:
-
Linkage disequilibrium
- ORs:
-
Odds ratios
- VSMC:
-
Vascular smooth muscle cells.
References
Pardali E, Goumans MJ, ten Dijke P: Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends Cell Biol. 2010, 20 (9): 556-567. 10.1016/j.tcb.2010.06.006.
Khan R, Agrotis A, Bobik A: Understanding the role of transforming growth factor-beta1 in intimal thickening after vascular injury. Cardiovasc Res. 2007, 74 (2): 223-234. 10.1016/j.cardiores.2007.02.012.
Schulick AH, Taylor AJ, Zuo W, Qiu CB, Dong G, Woodward RN, Agah R, Roberts AB, Virmani R, Dichek DA: Overexpression of transforming growth factor beta1 in arterial endothelium causes hyperplasia, apoptosis, and cartilaginous metaplasia. Proc Natl Acad Sci U S A. 1998, 95 (12): 6983-6988. 10.1073/pnas.95.12.6983.
Suthanthiran M, Li B, Song JO, Ding R, Sharma VK, Schwartz JE, August P: Transforming growth factor-beta 1 hyperexpression in African-American hypertensives: A novel mediator of hypertension and/or target organ damage. Proc Natl Acad Sci U S A. 2000, 97 (7): 3479-3484.
Majesky MW, Lindner V, Twardzik DR, Schwartz SM, Reidy MA: Production of transforming growth factor beta 1 during repair of arterial injury. J Clin Invest. 1991, 88 (3): 904-910. 10.1172/JCI115393.
Frutkin AD, Otsuka G, Stempien-Otero A, Sesti C, Du L, Jaffe M, Dichek HL, Pennington CJ, Edwards DR, Nieves-Cintron M, et al: TGF-[beta]1 limits plaque growth, stabilizes plaque structure, and prevents aortic dilation in apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2009, 29 (9): 1251-1257. 10.1161/ATVBAHA.109.186593.
Nikol S, Isner JM, Pickering JG, Kearney M, Leclerc G, Weir L: Expression of transforming growth factor-beta 1 is increased in human vascular restenosis lesions. J Clin Invest. 1992, 90 (4): 1582-1592. 10.1172/JCI116027.
Nabel EG, Shum L, Pompili VJ, Yang ZY, San H, Shu HB, Liptay S, Gold L, Gordon D, Derynck R, et al: Direct transfer of transforming growth factor beta 1 gene into arteries stimulates fibrocellular hyperplasia. Proc Natl Acad Sci U S A. 1993, 90 (22): 10759-10763. 10.1073/pnas.90.22.10759.
Grainger DJ, Kemp PR, Metcalfe JC, Liu AC, Lawn RM, Williams NR, Grace AA, Schofield PM, Chauhan A: The serum concentration of active transforming growth factor-beta is severely depressed in advanced atherosclerosis. Nat Med. 1995, 1 (1): 74-79. 10.1038/nm0195-74.
Wang XL, Sim AS, Wilcken DE: A common polymorphism of the transforming growth factor-beta1 gene and coronary artery disease. Clin Sci (Lond). 1998, 95 (6): 745-746. 10.1042/CS19980292.
Najar RA, Ghaderian SM, Panah AS: Association of Transforming Growth Factor-beta1 Gene Polymorphisms With Genetic Susceptibility to Acute Myocardial Infarction. Am J Med Sci. 2011, 342 (5): 365-370. 10.1097/MAJ.0b013e318215908a.
McCaffrey TA, Consigli S, Du B, Falcone DJ, Sanborn TA, Spokojny AM, Bush HL: Decreased type II/type I TGF-beta receptor ratio in cells derived from human atherosclerotic lesions. Conversion from an antiproliferative to profibrotic response to TGF-beta1. J Clin Invest. 1995, 96 (6)): 2667-2675.
Matt P, Schoenhoff F, Habashi J, Holm T, Van Erp C, Loch D, Carlson OD, Griswold BF, Fu Q, De Backer J, et al: Circulating transforming growth factor-beta in Marfan syndrome. Circulation. 2009, 120 (6): 526-532. 10.1161/CIRCULATIONAHA.108.841981.
Syrris P, Carter ND, Metcalfe JC, Kemp PR, Grainger DJ, Kaski JC, Crossman DC, Francis SE, Gunn J, Jeffery S, et al: Transforming growth factor-beta1 gene polymorphisms and coronary artery disease. Clin Sci (Lond). 1998, 95 (6): 659-667.
Yamada Y, Miyauchi A, Goto J, Takagi Y, Okuizumi H, Kanematsu M, Hase M, Takai H, Harada A, Ikeda K: Association of a polymorphism of the transforming growth factor-beta1 gene with genetic susceptibility to osteoporosis in postmenopausal Japanese women. J Bone Miner Res. 1998, 13 (10): 1569-1576.
Cambien F, Ricard S, Troesch A, Mallet C, Generenaz L, Evans A, Arveiler D, Luc G, Ruidavets JB, Poirier O: Polymorphisms of the transforming growth factor-beta 1 gene in relation to myocardial infarction and blood pressure. The Etude Cas-Temoin de l'Infarctus du Myocarde (ECTIM) Study. Hypertension. 1996, 28 (5)): 881-887.
Grainger DJ, Heathcote K, Chiano M, Snieder H, Kemp PR, Metcalfe JC, Carter ND, Spector TD: Genetic control of the circulating concentration of transforming growth factor type beta1. Hum Mol Genet. 1999, 8 (1): 93-97. 10.1093/hmg/8.1.93.
Shah R, Rahaman B, Hurley CK, Posch PE: Allelic diversity in the TGFB1 regulatory region: characterization of novel functional single nucleotide polymorphisms. Hum Genet. 2006, 119 (1–2): 61-74.
Dunning AM, Ellis PD, McBride S, Kirschenlohr HL, Healey CS, Kemp PR, Luben RN, Chang-Claude J, Mannermaa A, Kataja V, et al: A transforming growth factorbeta1 signal peptide variant increases secretion in vitro and is associated with increased incidence of invasive breast cancer. Cancer Res. 2003, 63 (10): 2610-2615.
Koch W, Hoppmann P, Mueller JC, Schomig A, Kastrati A: Association of transforming growth factor-beta1 gene polymorphisms with myocardial infarction in patients with angiographically proven coronary heart disease. Arterioscler Thromb Vasc Biol. 2006, 26 (5): 1114-1119. 10.1161/01.ATV.0000217747.66517.11.
Sie MP, Uitterlinden AG, Bos MJ, Arp PP, Breteler MM, Koudstaal PJ, Pols HA, Hofman A, van Duijn CM, Witteman JC: TGF-beta 1 polymorphisms and risk of myocardial infarction and stroke: the Rotterdam Study. Stroke. 2006, 37 (11): 2667-2671. 10.1161/01.STR.0000244779.30070.1a.
Crobu F, Palumbo L, Franco E, Bergerone S, Carturan S, Guarrera S, Frea S, Trevi G, Piazza A, Matullo G: Role of TGF-beta1 haplotypes in the occurrence of myocardial infarction in young Italian patients. BMC Med Genet. 2008, 9: 13-
Yokota M, Ichihara S, Lin TL, Nakashima N, Yamada Y: Association of a T29– > C polymorphism of the transforming growth factor-beta1 gene with genetic susceptibility to myocardial infarction in Japanese. Circulation. 2000, 101 (24): 2783-2787. 10.1161/01.CIR.101.24.2783.
Schunkert H, Konig IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, Preuss M, Stewart AF, Barbalic M, Gieger C, et al: Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011, 43 (4): 333-338. 10.1038/ng.784.
A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat Genet. 2011, 43 (4)): 339-344.
Sethupathy P, Collins FS: MicroRNA target site polymorphisms and human disease. Trends in genetics: TIG. 2008, 24 (10): 489-497. 10.1016/j.tig.2008.07.004.
Thakkinstian A, McEvoy M, Minelli C, Gibson P, Hancox B, Duffy D, Thompson J, Hall I, Kaufman J, Leung TF, et al: Systematic review and meta-analysis of the association between {beta}2-adrenoceptor polymorphisms and asthma: a HuGE review. Am J Epidemiol. 2005, 162 (3): 201-211. 10.1093/aje/kwi184.
Marchini J, Howie B: Genotype imputation for genome-wide association studies. Nat Rev Genet. 2010, 11 (7): 499-511. 10.1038/nrg2796.
Duval S, Tweedie R: Trim and fill: A simple funnel-plot-based method of testing and adjusting for publication bias in meta-analysis. Biometrics. 2000, 56 (2): 455-463. 10.1111/j.0006-341X.2000.00455.x.
Viechtbauer W: Conducting meta-analyses in R with the metafor package. Journal of Statistical Software. 2010, 36 (3): 1-48.
Wang Y, Luk AO, Ma RC, So WY, Tam CH, Ng MC, Yang X, Baum L, Lam V, Tong PC, et al: Independent predictive roles of eotaxin Ala23Thr, paraoxonase 2 Ser311Cys and beta-adrenergic receptor Trp64Arg polymorphisms on cardiac disease in Type 2 Diabetes–an 8-year prospective cohort analysis of 1297 patients. Diabet Med. 2010, 27 (4): 376-383. 10.1111/j.1464-5491.2010.02980.x.
Rao M, Guo D, Jaber BL, Tighiouart H, Pereira BJ, Balakrishnan VS: Transforming growth factor-beta 1 gene polymorphisms and cardiovascular disease in hemodialysis patients. Kidney Int. 2004, 66 (1): 419-427. 10.1111/j.1523-1755.2004.00748.x.
Manginas A, Tsiavou A, Chaidaroglou A, Giamouzis G, Degiannis D, Panagiotakos D, Cokkinos DV: Inflammatory cytokine gene variants in coronary artery disease patients in Greece. Coron Artery Dis. 2008, 19 (8): 575-582. 10.1097/MCA.0b013e32831286e8.
Oda K, Tanaka N, Arai T, Araki J, Song Y, Zhang L, Kuchiba A, Hosoi T, Shirasawa T, Muramatsu M, et al: Polymorphisms in pro- and anti-inflammatory cytokine genes and susceptibility to atherosclerosis: a pathological study of 1503 consecutive autopsy cases. Hum Mol Genet. 2007, 16 (6): 592-599.
Wang Y, Fu W, Xie F, Chu X, Wang H, Shen M, Sun W, Lei R, Yang L, Wu H, et al: Common polymorphisms in ITGA2, PON1 and THBS2 are associated with coronary atherosclerosis in a candidate gene association study of the Chinese Han population. J Hum Genet. 2010, 55 (8): 490-494. 10.1038/jhg.2010.53.
Sudomoina MA, Sukhinina TS, Barsova RM, Favorov AV, Shakhnovich RM, Titov BV, Matveeva NA, Rybalkin IN, Vlasik TN, Ochs MF, et al: Complex analsis of association of inflammation genes with myocardial infarction. Mol Biol (Mosk). 2010, 44 (3): 463-471.
Drenos F, Talmud PJ, Casas JP, Smeeth L, Palmen J, Humphries SE, Hingorani AD: Integrated associations of genotypes with multiple blood biomarkers linked to coronary heart disease risk. Hum Mol Genet. 2009, 18 (12): 2305-2316. 10.1093/hmg/ddp159.
Bobik A, Agrotis A, Kanellakis P, Dilley R, Krushinsky A, Smirnov V, Tararak E, Condron M, Kostolias G: Distinct patterns of transforming growth factor-beta isoform and receptor expression in human atherosclerotic lesions, Colocalization implicates TGF-beta in fibrofatty lesion development. Circulation. 1999, 99 (22): 2883-2891. 10.1161/01.CIR.99.22.2883.
Piao M, Tokunaga O: Significant expression of endoglin (CD105), TGFbeta-1 and TGFbeta R-2 in the atherosclerotic aorta: an immunohistological study. J Atheroscler Thromb. 2006, 13 (2): 82-89. 10.5551/jat.13.82.
Panutsopulos D, Papalambros E, Sigala F, Zafiropoulos A, Arvanitis DL, Spandidos DA: Protein and mRNA expression levels of VEGF-A and TGF-beta1 in different types of human coronary atherosclerotic lesions. Int J Mol Med. 2005, 15 (4): 603-610.
O'Brien ER, Bennett KL, Garvin MR, Zderic TW, Hinohara T, Simpson JB, Kimura T, Nobuyoshi M, Mizgala H, Purchio A, et al: Beta ig-h3, a transforming growth factor-beta-inducible gene, is overexpressed in atherosclerotic and restenotic human vascular lesions. Arterioscler Thromb Vasc Biol. 1996, 16 (4): 576-584. 10.1161/01.ATV.16.4.576.
McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, Hinds DA, Pennacchio LA, Tybjaerg-Hansen A, Folsom AR, et al: A common allele on chromosome 9 associated with coronary heart disease. Science. 2007, 316 (5830): 1488-1491. 10.1126/science.1142447.
Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, Jonasdottir A, Sigurdsson A, Baker A, Palsson A, et al: A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007, 316 (5830): 1491-1493. 10.1126/science.1142842.
Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, Dixon RJ, Meitinger T, Braund P, Wichmann HE, et al: Genomewide association analysis of coronary artery disease. N Engl J Med. 2007, 357 (5): 443-453. 10.1056/NEJMoa072366.
Angelakopoulou A, Shah T, Sofat R, Shah S, Berry DJ, Cooper J, Palmen J, Tzoulaki I, Wong A, Jefferis BJ, et al: Comparative analysis of genome-wide-association studies signals for lipids, diabetes, and coronary heart disease: Cardiovascular Biomarker Genetics Collaboration. Eur Heart J. 2011, 3: 1-2.
Glagov S, Weisenberg E, Zarins CK, Stankunavicius R, Kolettis GJ: Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med. 1987, 316 (22): 1371-1375. 10.1056/NEJM198705283162204.
McPherson DD, Sirna SJ, Hiratzka LF, Thorpe L, Armstrong ML, Marcus ML, Kerber RE: Coronary arterial remodeling studied by high-frequency epicardial echocardiography: an early compensatory mechanism in patients with obstructive coronary atherosclerosis. J Am Coll Cardiol. 1991, 17 (1): 79-86. 10.1016/0735-1097(91)90707-G.
Stiel GM, Stiel LS, Schofer J, Donath K, Mathey DG: Impact of compensatory enlargement of atherosclerotic coronary arteries on angiographic assessment of coronary artery disease. Circulation. 1989, 80 (6): 1603-1609. 10.1161/01.CIR.80.6.1603.
Hermiller JB, Tenaglia AN, Kisslo KB, Phillips HR, Bashore TM, Stack RS, Davidson CJ: In vivo validation of compensatory enlargement of atherosclerotic coronary arteries. Am J Cardiol. 1993, 71 (8): 665-668. 10.1016/0002-9149(93)91007-5.
Schoenhagen P, Ziada KM, Kapadia SR, Crowe TD, Nissen SE, Tuzcu EM: Extent and direction of arterial remodeling in stable versus unstable coronary syndromes: an intravascular ultrasound study. Circulation. 2000, 101 (6): 598-603. 10.1161/01.CIR.101.6.598.
Okura H, Kobayashi Y, Sumitsuji S, Terashima M, Kataoka T, Masutani M, Ohyanagi M, Shimada K, Taguchi H, Yasuga Y, et al: Effect of culprit-lesion remodeling versus plaque rupture on three-year outcome in patients with acute coronary syndrome. Am J Cardiol. 2009, 103 (6): 791-795. 10.1016/j.amjcard.2008.11.030.
Fischer M, Broeckel U, Holmer S, Baessler A, Hengstenberg C, Mayer B, Erdmann J, Klein G, Riegger G, Jacob HJ, et al: Distinct heritable patterns of angiographic coronary artery disease in families with myocardial infarction. Circulation. 2005, 111 (7): 855-862. 10.1161/01.CIR.0000155611.41961.BB.
Nishioka T, Nagai T, Luo H, Kitamura K, Hakamata N, Akanuma M, Katsushika S, Uehata A, Takase B, Isojima K, et al: Coronary remodeling of proximal and distal stenotic atherosclerotic plaques within the same artery by intravascular ultrasound study. Am J Cardiol. 2001, 87 (4): 387-391. 10.1016/S0002-9149(00)01388-6.
Helgadottir A, Thorleifsson G, Magnusson KP, Gretarsdottir S, Steinthorsdottir V, Manolescu A, Jones GT, Rinkel GJ, Blankensteijn JD, Ronkainen A, et al: The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet. 2008, 40 (2): 217-224. 10.1038/ng.72.
Biros E, Cooper M, Palmer LJ, Walker PJ, Norman PE, Golledge J: Association of an allele on chromosome 9 and abdominal aortic aneurysm. Atherosclerosis. 2010, 212 (2): 539-542. 10.1016/j.atherosclerosis.2010.06.015.
Thompson AR, Golledge J, Cooper JA, Hafez H, Norman PE, Humphries SE: Sequence variant on 9p21 is associated with the presence of abdominal aortic aneurysm disease but does not have an impact on aneurysmal expansion. Eur J Hum Genet. 2009, 17 (3): 391-394. 10.1038/ejhg.2008.196.
Yasuno K, Bilguvar K, Bijlenga P, Low SK, Krischek B, Auburger G, Simon M, Krex D, Arlier Z, Nayak N, et al: Genome-wide association study of intracranial aneurysm identifies three new risk loci. Nat Genet. 2010, 42 ((5): 420-425.
Nakaoka H, Takahashi T, Akiyama K, Cui T, Tajima A, Krischek B, Kasuya H, Hata A, Inoue I: Differential effects of chromosome 9p21 variation on subphenotypes of intracranial aneurysm: site distribution. Stroke. 2010, 4 (8): 1593-1598.
Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, et al: Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006, 312 (5770): 117-121. 10.1126/science.1124287.
Shimizu C, Jain S, Davila S, Hibberd ML, Lin KO, Molkara D, Frazer JR, Sun S, Baker AL, Newburger JW, et al: Transforming growth factor-beta signaling pathway in patients with Kawasaki disease. Circ Cardiovasc Genet. 2011, 4 (1): 16-25. 10.1161/CIRCGENETICS.110.940858.
Pannu H, Fadulu VT, Chang J, Lafont A, Hasham SN, Sparks E, Giampietro PF, Zaleski C, Estrera AL, Safi HJ, et al: Mutations in transforming growth factor-beta receptor type II cause familial thoracic aortic aneurysms and dissections. Circulation. 2005, 112 (4): 513-520. 10.1161/CIRCULATIONAHA.105.537340.
Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, et al: A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005, 37 (3): 275-281. 10.1038/ng1511.
van de Laar IM, Oldenburg RA, Pals G, Roos-Hesselink JW, de Graaf BM, Verhagen JM, Hoedemaekers YM, Willemsen R, Severijnen LA, Venselaar H, et al: Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet. 2011, 43 (2): 121-126. 10.1038/ng.744.
Prakash SK, LeMaire SA, Guo DC, Russell L, Regalado ES, Golabbakhsh H, Johnson RJ, Safi HJ, Estrera AL, Coselli JS, et al: Rare copy number variants disrupt genes regulating vascular smooth muscle cell adhesion and contractility in sporadic thoracic aortic aneurysms and dissections. Am J Hum Genet. 2010, 87 (6): 743-756. 10.1016/j.ajhg.2010.09.015.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/13/39/prepub
Acknowledgments
We thank Professor Philippa J. Talmud for sharing data with us. This work was supported by grant 2006B195 of the Netherlands Heart Foundation, and partially (RMB and OF) by grant 8/3-280n-10 of the Moscow Government. See Additional file 1 for the acknowledgments for the PROCARDIS study and the CARDIoGRAM study.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YL, JMB, RMB, OF, AG, CARDIoGRAM Consortium, MM, and EJF have made substantial contributions to conception and design, or acquisition of the data, or analysis and interpretation of data; YL, JMB, RMB, OF, and EJF have been involved in drafting the manuscript or revising it critically for important intellectual content; and all authors have given final approval of the version to be published. All authors read and approved the final manuscript.
Members and affiliations of the CARDIoGRAM Consortium are listed in the Additional files.
Electronic supplementary material
12881_2011_987_MOESM2_ESM.doc
Additional file 2: Table S1. Summary of the meta-analysis of studies examining the association between TGFB1 polymorphisms and coronary heart disease risk. (DOC 38 KB)
12881_2011_987_MOESM4_ESM.doc
Additional file 4: Figure S1. Funnel plots with pseudo 95% confidence intervals for rs1800469 analysed according to different genotype contrasts. A. Comparison of the homozygous TT genotype with the wild type CC genotype (fixed-effect model, p for Egger’s regression test = 0.05); B. Comparison of the heterozygous CT genotype with the wild type CC genotype (fixed-effect model, p for Egger’s regression test = 0.61); C. Comparison of the TT+CT genotype with the wild type CC genotype (fixed-effect model, p for Egger’s regression test = 0.22). Figure S2. Funnel plots with pseudo 95% confidence intervals for rs1982073 analysed according to different genotype contrasts. A. Comparison of the homozygous CC genotype with the wild type TT genotype (random-effect model, p for Egger’s regression test = 0.94); B. Comparison of the heterozygous TC genotype with the wild type TT genotype (random-effect model, p for Egger’s regression test = 0.50); C. Comparison of the variant genotype of CC+TC with the wild type TT genotype (random-effect model, p for Egger’s regression test = 0.71). Figure S3. Funnel plots with pseudo 95% confidence intervals for rs1800471 analysed according to different genotype contrasts. A. Comparison of the homozygous CC genotype with the wild type GG genotype (fixed-effect model, p for Egger’s regression test = 0.75); B. Comparison of the heterozygous GC genotype with the wild type GG genotype (fixed-effect model, p for Egger’s regression test = 0.89); C. Comparison of the variant genotype of CC+GC with the wild type GG genotype (fixed-effect model, p for Egger’s regression test = 0.83). (DOC 54 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lu, Y., Boer, J.M., Barsova, R.M. et al. TGFB1 genetic polymorphisms and coronary heart disease risk: a meta-analysis. BMC Med Genet 13, 39 (2012). https://doi.org/10.1186/1471-2350-13-39
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-13-39