
Abstract
Density functional theory calculations have been conducted to explore the electronic structure of bimetallic nanoclusters X12Y (X = Pt, Cu; Y = Ag, Al, Au, Bi, Cu, Ni, Pt, Ti). The results demonstrate that the properties of metal clusters can be tuned by dopant atoms.
Similar content being viewed by others
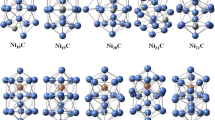
References
B. R. Cuenya, “Synthesis and catalytic properties of metal nanoparticles: size, shape, support, composition, and oxidation state effects,” Thin Solid Films 518, 3127–3150 (2010)
V. I. Bukhtiyarov and M. G. Slin’ko, “Metallic nano- systems in catalysis,” Usp. Khim. 70(2), 167 (2001).
R. Ferrando, K. Jellinek, and R. L. Johnston, “Nanoalloys: from theory to applications of alloy clusters and nanoparticles,” Chem. Rev. 108(3), 846 (2008).
T. Ozaki, “Variationally optimized atomic orbitals for large-scale electronic structures,” Phys. Rev. B 67,155108 (2003).
T. Ozaki and H. Kino, “Numerical atomic basis orbitals from H to Kr,” Phys. Rev. 69, 195113 (2004).
I. Morrison, D. M. Bylander, and L. Kleinman, “Non-local Hermitian norm-conserving Vanderbild pseudo-potential,” Phys. Rev. B 47, 6728 (1993).
J. P. Perdew, K. Burke, and M. Ernzerhof, “Generalized gradient approximation made simple,” Phys. Rev. Lett. 77, 3865 (1996).
R. A. Olsen, P. H. T. Philipsen, and E. J. Baerends, “CO on Pt(111): a puzzle revisited,” J. Chem. Phys. 119(8), 4522 (2003).
B. Hammer, O. H. Nielsen and J. K. Norskov, “Structure sensitivity in adsorption: CO interaction with stepped and reconstructed Pt surfaces,” Catal. Lett. 46, 31–35 (1997).
L.-L. Wang and D. D. Johnson, “Electrocatalytic properties of PtBi and PtPb intermetallic line compounds via DFT: CO and H adsorption,” J. Phys. Chem. 112, 8266 (2008).
M. Mavrikakis, B. Hammer, and J. K. Norskov, “Effect of strain on the reactivity of metal surfaces,” Phys. Rev. Lett. 81(13), 2819 (1998).
B. H. Morrow, D. E. Resasco, A. Striolo, and M. B. Nardelli, “CO adsorption on noble metal clusters: local environment effects,” J. Phys. Chem. C 115, 5637 (2011).
E. Knoesel, A. Hotzel, and M. Wolf, “Ultrafast dynamics of hot electrons and holes in copper: excitation, energy relaxation, and transport effects,” Phys. Rev. B 57, 12812 (1998).
W. Tang and G. Henkelman, “Charge redistribution in core-shell nanoparticles to promote oxygen reduction,” J. Chem. Phys. 130, 194504 (2009).
Author information
Authors and Affiliations
Corresponding author
Additional information
Original Russian Text © N.N. Kolchenko, N.A. Chernyshev, 2013, published in Rossiiskie Nanotekhnologii, 2013, Vol. 8, Nos. 7–8.
Rights and permissions
About this article
Cite this article
Kolchenko, N.N., Chernyshev, N.A. Electronic structure and adsorption property of doped metal clusters. Nanotechnol Russia 8, 445–451 (2013). https://doi.org/10.1134/S199507801304006X
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S199507801304006X