
Abstract
Determining the cellular localization of proteins of interest at nanometer resolution is necessary for elucidating their functions. Besides super-resolution fluorescence microscopy, conventional electron microscopy (EM) combined with immunolabeling or clonable EM tags provides a unique approach to correlate protein localization information and cellular ultrastructural information. However, there are still rare cases of such correlation in three-dimensional (3D) spaces. Here, we developed an approach by combining the focus ion beam scanning electron microscopy (FIB-SEM) and a promising clonable EM tag APEX2 (an enhanced ascorbate peroxidase 2) to determine the target protein localization within 3D cellular ultrastructural context. We further utilized this approach to study the 3D localization of mitochondrial dynamics-related proteins (MiD49/51, Mff, Fis1, and Mfn2) in the cells where the target proteins were overexpressed. We found that all the target proteins were located at the surface of the mitochondrial outer membrane accompanying with mitochondrial clusters. Mid49/51, Mff, and hFis1 spread widely around the mitochondrial surface while Mfn2 only exists at the contact sites.
Similar content being viewed by others

Introduction
Protein localization correlates with its particular function in cells or tissues. Mapping protein localization information onto their cellular ultrastructural context is of great importance for cell biology study and can be achieved via electron microscopy (EM). Generally, there are two ways to localize a target protein in EM: immune-localization and clonable tags localization. The EM contrast of immune-localization comes from the antibody-conjugated gold particles (De Mey et al. 1981) or quantum dots (Giepmans et al. 2005). This approach is significantly limited due to the limited efficiency of immunolabeling, the spatial hindrance of large antibodies, and the fact that the well-preserved ultrastructure and the antigen immuno-activity are always mutually exclusive.
Clonable tags localization has been achieved via metallothionein, a small cysteine-rich protein that can bind a variety of heavy metal ions with its cysteine residues (Hamer 1986; Kagi and Schaffer 1988; Mercogliano and DeRosier 2007). To avoid the possible heavy metal toxicity or its influence on the behavior of target proteins, the cells were treated with the heavy metal during EM sample preparation (Morphew et al. 2015). However, the preservation of the cellular ultrastructure and the low tolerance of metallothionein to strong chemical fixation are still mutually exclusive. Another kind of clonable tag is based on the oxidization of 3,3′-diaminobenzidine (DAB). The contrast comes from the enriched osmium tetroxide that is recruited by the osmiophilic DAB polymer. According to the type of oxidation, these tags can be further classified into photo-oxidation tags, which include fluorescent protein (FP) (Grabenbauer et al. 2005; Meißlitzer-Ruppitsch et al. 2008), resorufin arsenical hairpin (ReAsH) (Gaietta et al. 2002), and mini singlet oxygen generator (miniSOG) (Shu et al. 2011), or they can be classified as enzyme-based oxidation tags, which include horseradish peroxidase (HRP) (Connolly et al. 1994; Li et al. 2010) and enhanced ascorbate peroxidase (APEX/APEX2) (Martell et al. 2012; Lam et al. 2015 ). Considering the low-yield efficiency of singlet oxygen by FPs as well as the relative low EM contrast, the usage of FPs is limited (Su et al. 2010). ReAsH works based on the reaction between tetracysteine tag and biarsenical compounds (Adams et al. 2002), which yields a problem of nonspecific labeling. Moreover, one general concern for the photo-oxidation-based tags is the inaccessibility of light in a deep tissue. Although the enzyme-based tags take advantage in dealing with thick tissue, the targeting capabilities of HRP in cytosol limit their usage (Hopkins et al. 2000). However, APEX/APEX2 has become a promising clonable EM tag because it can not only preserve cellular ultrastructure well but also give excellent EM contrast, regardless of the thickness of the specimen and the localization of target proteins (Martell et al. 2012). It is important to note that APEX2 is an A134P mutant of APEX with improved kinetics, thermal stability, heme binding, and resistance to high H2O2 concentrations (Lam et al. 2015).
Volume electron microscopy technique can study cellular ultrastructure in three dimensions (3D), which provides more information compared to the 2D projection or single slice. The transmission electron microscopy (TEM)-based volume EM methods include electron tomography (ET) (Hart 1968; Koster et al. 1992), serial sectioning followed by TEM (ssTEM) (Harris et al. 2006), and serial section ET (Ladinsky et al. 1999). The scanning electron microscopy (SEM)-based volume EM methods include serial section SEM (ssSEM) (Horstmann et al. 2012), serial block face scanning electron microscopy (SBF-SEM) (Denk and Horstmann 2004), and focus ion beam scanning electron microscopy (FIB-SEM) (Knott et al. 2008). For a small volume (50 × 50 × 30 μm3), FIB-SEM approach provides an efficient and automatic way to get the 3D ultrastructure of specimen with a higher axial resolution in comparison to serial section-based techniques and SBF-SEM (Peddie and Collinson 2014; Li et al. 2016).
As a highly dynamic organelle in eukaryotic cells, mitochondria perpetually divide, fuse, and move in response to ever-changing physiological demand of the cells (Youle and Van Der Bliek 2012). There are many proteins involved in mitochondrial dynamics and regulation. In mammalian cells, mitochondrial dynamics proteins 49 and 51 kDa (MiD49 and MiD51, respectively) (Palmer et al. 2011), fission protein 1 (Fis1, and hFis1 for human homologues) (Yoon et al. 2003), and mitochondrial fission factor (Mff) (Otera et al. 2010) are mitochondrial outer membrane proteins and play an important role as receptors of dynamic-related protein 1 (Drp1), which drives mitochondrial fission (Smirnova et al. 2001). Mitofusin 1 and 2 (Mfn1 and Mfn2, respectively) control the fusion of mitochondrial outer membrane (Santel and Fuller 2001) while OPA1 in both short and long forms play the central role of mitochondrial inner membrane fusion (Song et al. 2007). Overexpression of these proteins in mammalian cells will induce significant clustering of mitochondria, which was revealed from fluoresce microscopy (Griparic et al. 2004; Huang et al. 2007; Otera et al. 2010; Zhao et al. 2011; Liu et al. 2013). However, there was no information available on how these proteins are localized within the cluster and how these clustered mitochondria look like in 3D space.
In the present study, we developed a 3D localization EM method (APEX2–FIB-SEM) by combining FIB-SEM and APEX2 techniques to enable the correlation of protein localization information and cellular ultrastructure in the 3D space of a single cell with a high resolution. We utilized this approach to study the 3D localization of mitochondrial fusion- and fission-related proteins, including MiD51, MiD49, hFis1, Mff, and Mfn2.
Results
Workflow of APEX2–FIB-SEM
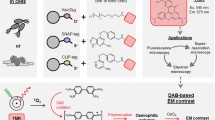
The gene of the target protein with APEX2 fused at the N or C terminus was cloned into an appropriate vector (e.g., pcDNA3 here) for transfection. The cells (e.g., HEK 293T cells here) were cultured on a sterile plastic coverslip within a culture dish (Fig. 1A). After transfection and further culturing, the cells were fixed, stained with DAB, dehydrated, and then embedded in resin as previously reported (Fig. 1B) (Martell et al. 2012). Considering the reproducibility, we recommend to use the commercial DAB Kit (e.g., CWBIO here) for DAB staining. Various embedding molds can be used for the polymerization of the resin-embedded cells with coverslip as long as they can be separated easily without damaging the cells (Fig. 1C). After polymerization, the plastic coverslip was removed carefully and a few ultrathin sections from the surface of the resin block were checked by TEM to screen for the optimally stained sample with well-preserved ultrastructure that will be further imaged by FIB-SEM. Only three to five slices were sectioned from the surface of the resin block and the thickness of each section was less than 80 nm to make sure there were still enough samples for subsequent FIB-SEM experiments (Fig. 1D). The details of the workflow are described in “Materials and methods”.

The scheme for APEX2–FIB-SEM. A The cells are cultured on sterile plastic coverslips in dishes, and transfected with the target genes fused with APEX2. The cells on the plastic coverslip are used for further EM sample preparation. B The cells on the plastic coverslip undergo fixation, DAB staining, dehydration, and resin embedment. C Before polymerization, the plastic coverslip with cells is transferred to the dish placed with aluminum foil. The cell side of the plastic coverslip is facing up. Fresh resin with accelerator is added to cover the cells. D After polymerization, the block is separated from aluminum foil and plastic coverslip, and the areas with staining patterns are cut off and stuck on the tip of an empty resin block. In order to reserve enough sample for FIB-SEM (colored with green), only three to five slices (less than 80 nm) are sectioned from the block (colored with blue) for TEM screening
3D Localization of Mitochondrial Dynamics-Related Proteins
We first utilized mitochondrial calcium uniporter (MCU) as a positive control that was used in the previous APEX study (Martell et al. 2012) to validate the performance of APEX/APEX2 staining in our system. In our TEM experiment, MCU-APEX gave a strong EM contrast within mitochondrial matrix (Fig. 2A), which was consistent with the previous report (Martell et al. 2012) and showed that APEX worked well in our workflow.

Visualizing the staining patterns in 2D by TEM. The micrographs show cells expressing MCU-APEX (A), MiD51-APEX2 (B), MiD49-APEX2 (C), APEX2-Mff (D), APEX2-hFis1 (E), and APEX2-Mfn2 (F). The staining areas with strong EM contrast indicate the locations of the target proteins (black arrows). The labels in the top right corner of each micrograph show the relevant position between the target protein and the tag as well as their relevant sizes. Scale bars, 1 μm
In our further experiments, we selected APEX2 instead of APEX since APEX2 was more active due to its better resistance to high H2O2 concentration (Lam et al. 2015). By examining ultrathin sections in TEM, we found that the constructs MiD51-APEX2 (Fig. 2B), MiD49-APEX2 (Fig. 2C), APEX2-Mff (Fig. 2D), APEX2-hFis1 (Fig. 2E), and APEX2-Mfn2 (Fig. 2F) could yield a good EM contrast at the reasonable positions. These constructs were then selected for the subsequent FIB-SEM experiments. It is to be noted that, for the constructs Mff-APEX2, hFis1-APEX2, and Mfn2-APEX2, we could not find reasonable staining patterns in TEM micrographs. We speculated that it is due to the fusion of APEX2 in the proximity of the transmembrane helices of the target proteins, which induced the failure of their proper localizations in mitochondrial outer membrane (see also in “Discussions”).
MiD51/49 has been found to recruit Drp1 into mitochondria, and overexpression of MiD51 or MiD49 induced mitochondrial elongation, which was thought to be the phenotype of mitochondrial fusion (Zhao et al. 2011; Liu et al. 2013). We also consistently found that mitochondria appeared as elongated tubulars or compact clusters in the cells overexpressing MiD51/49-APEX2 (Figs. 2B, C and 3A, B). However, the mitochondrial fusion phenotype induced by overexpression of MiD51/49 varied from cells to cells by the comparison of the EM images of cells expressing MiD51-APEX2 (see Figs. 2B and 3A) or those expressing MiD49-APEX2 (see Figs. 2C and 3B). We therefore speculated that the mitochondrial fusion phenotype induced by MiD51/MiD49 is highly dependent on their expression level (see also in “Discussions”). Through investigation of the 3D volume of the clustered mitochondria (Fig. 3A and B; see also Movies S1 and S2), we clearly found that the target proteins MiD51/MiD49 were largely localized at the interface among mitochondria with reduced amount of proteins surrounding the peripheral of the mitochondrial cluster. This observation implied how MiD51/MiD49 induced mitochondrial clustering by recruiting Drp1.

Visualizing the staining pattern in 3D by FIB-SEM. The figure panels show cells expressing MiD51-APEX2 (A), MiD49-APEX2 (B), APEX2-Mff (C), APEX2-Fis1 (D), and APEX2-Mfn2 (E). The left columns display the representative SEM micrographs from each volume of EM data, and the right columns display the 3D rendering of boxed areas of the left with the yellow spheres for mitochondria and purple layers for APEX2-induced EM contrast. The labels in the top left corner of each micrograph show the relevant position between the target protein and the tag as well as their relevant sizes. Scale bars, 5 μm
Mff was thought to be an essential factor for mitochondrial recruitment of Drp1 during mitochondria fission (Otera et al. 2010). In the cells with exogenous expression of APEX2-Mff, the mitochondria appeared in circular shapes and formed compact clusters (Figs. 2D and 3C) in consistency with the previous finding (Otera et al. 2010). Through investigation of the 3D volume of the clustered mitochondria, we clearly found that Mff was not only rich in the areas of mitochondrial connection, but also presented at the periphery of the mitochondrial cluster (Fig. 3C; see also Movie S3).
Fis1 was the first proposed Drp1 receptor and could induce the mitochondrial fission (Mozdy et al. 2000, James et al. 2003, Yoon et al. 2003). In the APEX2-hFis1-transfected cells, the clusters of vesicular mitochondria were found (Figs. 2E and 3D) in consistency with the previous study (Otera et al. 2010). The clustering phenotype also possibly varied in an expression level-dependent manner from cells (Fig. 2E) to cells (Fig. 3D). Through investigation of the 3D volume of the clustered mitochondria, we also found that hFis1 was located in high density at the interface of clustered mitochondria with a significant amount at the periphery of mitochondrial cluster (Fig. 3D; see also Movie S4).
Mfn2 can mediate tethering and fusion of mitochondrial outer membrane during the mitochondrial fusion process (Koshiba et al. 2004; Meeusen et al. 2004), with both of its N-terminal and C-terminal domains exposed towards the cytosol (Rojo et al. 2002). In the cells overexpressing APEX2-Mfn2, the mitochondria formed clusters and most of mitochondria were in circular shapes (Figs. 2F and 3E), which was consistent with the previous description of grape-like aggregation (Huang et al. 2007). However, to our surprise, unlike the clusters induced by the four proteins above, here, the mitochondrial cluster induced by overexpressing APEX2-Mfn2 was relatively loose, especially at the inside region that was filled with strong staining signals. In addition, through investigation into the 3D volume of the clustered mitochondria, we found that Mfn2 was only located in the contact sites of clustered mitochondria, and no staining signal was found at the periphery of mitochondrial cluster (Fig. 3E; see also Movie S5). This finding was significantly different from those associated with mitochondrial fission-related proteins MiD51, MiD49, Mff, and hFis1.
Discussion
In this study, we developed the APEX2–FIB-SEM approach to determine the target protein localization within 3D cellular ultrastructural context by combining two state-of-the-art techniques, APEX2 and FIB-SEM. By using APEX2–FIB-SEM, we successfully mapped the position of mitochondrial dynamics-related proteins (MiD51/49, Mff, hFis1, and Mfn2) when they were overexpressed. The clusters with tubular (MiD51/49) or circular (Mff, hFis1, and Mfn2) mitochondria were found in those cells. Through investigation of the staining patterns in the 3D volume data, we found Mff and hFis1 distributed widely around the surface of mitochondrial outer membrane, and they were consistent with MiD51/49 except that the staining signals of MiD51/49 were reduced at the periphery of mitochondrial cluster. However, the relatively loose mitochondrial clusters and extremely restrained staining patterns at the mitochondrial contact site suggested that the mechanism of the fusion phenotype induced by overexpressing Mfn2 was distinctive from that of fission factors.
For alpha-helically anchored mitochondrial outer membrane proteins, the targeting signal is confined to transmembrane domain and positive residues in its flanking regions (McBride et al. 1992; Waizenegger et al. 2003; Bruggisser et al. 2017). The failure of the constructs, such as Mff-APEX2, hFis1-APEX2, and Mfn2-APEX2, is probably due to the fusion of APEX2 to the terminal which is close to the transmembrane region, and perturbs their translocation.
Like tagged with APEX2, similar morphologies are present in cells overexpressing the above five proteins without APEX2 (Huang et al. 2007; Otera et al. 2010; Zhao et al. 2011; Liu et al. 2013), which indicates that these mitochondrial phenotypes are not tag-associated artifacts. Their ubiquitous distributions at the interface regions of clustered mitochondria suggest that these proteins participate in tethering the mitochondria together at the outer membrane surface.
The different expression levels of the exogenous genes may induce distinct phenotype of cells and different behavior of proteins. As a result, the comparison of cellular phenotypes and location of proteins should be made with approximate expression levels. With too high expression level, the EM contrast would appear at unreasonable areas, for example in Fig. 3A, the MiD51 directed staining is also found at cytosol. The dispersing signals may result from the accumulated mis-located fusion proteins in cytosol.
In addition, the endogenous oxidases can also polymerize DAB and give false-positive signals, such as the cytochrome oxidase that is located in the intermembrane space of mitochondria (Seligman et al. 1968). However, most of the endogenous oxidases should have lower tolerance to strong chemical fixative than APEX/APEX2, which is the prerequisite to ensure APEX/APEX2 can work without interruption from endogenous oxidases. As a result, the fixation level should be well regulated to ensure that the endogenous oxidases are fully inactive without disrupting the activity of APEX/APEX2. In our experiment, the detached and pelleted cells were also tested for DAB staining and EM sample preparation, and uninterpretable staining was found at the mitochondrial intermembrane space and endosome matrix (data not shown). Moreover, staining was also found in the cells without transfection. However, the false-positive staining was missing with the same procedures for the monolayer cells. So the protocol of fixation and staining in this study is suitable for monolayer cells, and slight modification should be done in the fixation step for other kinds of samples.
Materials and Methods
Plasmids
All the genes were cloned into the pcDNA3.0 vector with five Myc tags near 3′ end of the multiple cloning site, respectively. All of them were inserted before c-Myc tag with a terminator. The A134P point mutation of APEX which turned APEX to APEX2 was created using extension overlap PCR. MiD51/49 and MCU were fused at the N-terminal of the APEX/APEX2, and Mff, hFis1, and Mfn2 were fused at both N- and C-terminals of the APEX2. MiD51-APEX2, MiD49-APEX2, hFis1-APEX2, Mfn2-APEX2, and MCU-APEX were cloned between BamH I and EcoR I sites. Mff-APEX2, APEX2-Mff, APEX2-hFis1, and APEX2-Mfn2 were cloned into Kpn I and BamH I sites. These fused genes were generated by overlap extension PCR, and the linker sequence between them was 5′-CTGGACAGCACC-3′. The fused genes were inserted into vector using standard restriction cloning methods and verified by nucleotide sequencing.
Cell Culture and Transfection
HEK 293T cells were cultured in Dulbecco’s modified eagle medium (DMEM, Corning) supplemented with 10% (v/v) fetal bovine serum (Gibco). The cultures were kept in the condition of 5% CO2 and 37 °C. After culturing, the cells were placed into a 12-well plate (Corning) with each well having 200,000 cells approximately. It is to be noted that, before plating cells, a resized sterile plastic coverslip (Thermanox) was placed on the bottom of each well. After additional 24 h culturing, the cells were transfected by X-treme HP (Roche technologies), and then fixed after another 48–72 h.
Sample Preparation for Electron Microscopy
The DAB staining and EM sample preparation were as previously reported (Martell et al. 2012) with slight modification. The plastic coverslip with transfected cells were fixed by 2.5% glutaraldehyde (Electron Microscopy Science) for 1 h on ice. Then they were washed by phosphate-buffered solution (PBS, pH 7.4) three times. Then the cells were incubated at 20 mmol/L glycine in PBS for 5 min, and rinsed by chilled PBS 3 × 5 min. DAB Kit (CWBIO) was used to stain cells for 15 min, followed by 3 × 2 min rinses in chilled PBS. The formation of DAB polymer could be checked by transmission light microscope. The cells were further post-fixed and stained by 2% osmium tetroxide (Electron Microscopy Science) for 30 min, and rinsed by distilled water 3 × 5 min. Then 2% aqueous uranyl acetate (Electron Microscopy Sciences) was placed on cells overnight. Thereafter, cells were gradient-dehydrated in ethanol series (30%, 50%, 70%, 80%, 95%, 100%) and 100% acetone. During the dehydration process of using 100% acetone, cells were transferred from ice temperature to room temperature. Cells were embedded by diluted Epon 812 (Electron Microscopy Sciences) in acetone (25%, 50%, 75%) for 2 h each and then by 100% resin three times with each time for 2 h. Before the resin polymerization step, the plastic coverslip with cells was transferred to a six-well plate coated with aluminum foil: the aluminum foil on the bottom of plate needs to be as flat as possible. The surface of plastic coverslip with cells was upward, and fresh resin with accelerator was added into the plates to cover the cells. The polymerization was performed at 60 °C for 48 h. Then the blocks could be taken out from the six-well plate and carefully separated from the aluminum foil and plastic coverslip. The cells should be near the surface of the blocks. Before the following steps, the block could be observed under a light microscope and the areas with significant staining signals should be cut off and stuck on the tip of a blank resin block that was polymerized in standard flat embedding mold (Electron Microscopy Sciences).
Transmission Electron Microscopy
The blocks were trimmed and sectioned into 80 nm using an ultramicrotome (Leica EM UC6). Electron micrographs were recorded using FEI Tecnai spirit TEM (Thermo Fisher Scientific) operated at 120 kV.
FIB-SEM and Image Processing
The block with the target regions was glued onto a 45° inclined aluminum sample stub with conductive silver paint. To avoid the charging artifacts, the blocks were coated with a thin-layer of carbon. The data were collected by FEI Helios NanoLab 600i Dual beam SEM (Thermo Fisher Scientific). The platinum layer was deposited on the region of interest with a gas injection system to protect the specimen surface from ion beam-induced damage. The accelerating voltage of FIB was 30 kV, and the beam current was set to 0.79 nA with the milling thickness of 15 or 20 nm for different volume data. The series micrographs were recorded using an accelerating voltage of 2 kV, a current of 0.69 nA, and a dwell time of 8 μs. The FIB-SEM data were aligned, reconstructed, and segmented by the Amira software (Thermo Fisher Scientific).
Abbreviations
- 3D:
-
Three dimensions
- APEX:
-
Enhanced ascorbate peroxidase
- CMV:
-
Cytomegalovirus
- DAB:
-
3,3′- diaminobenzidine
- Drp1:
-
Dynamic-related protein 1
- EM:
-
Electron microscopy
- ET:
-
Electron tomography
- FIB-SEM:
-
Focus ion beam scanning electron microscopy
- Fis1:
-
Fission protein 1
- FP:
-
Fluorescent protein
- HRP:
-
Horseradish peroxidase
- MCU:
-
Mitochondrial calcium uniporter
- Mff:
-
Mitochondrial fission factor
- Mfn:
-
Mitofusin
- MiD:
-
Mitochondrial dynamics protein
- miniSOG:
-
Mini singlet oxygen generator
- ReAsH:
-
Resorufin arsenical hairpin
- ssTEM:
-
Serial section transmission electron microscopy
- ssSEM:
-
Serial section scanning electron microscopy
- SBF-SEM:
-
Serial block face scanning electron microscopy
- TEM:
-
Transmission electron microscopy
References
Adams SR, Campbell RE, Gross LA, Martin BR, Walkup GK, Yao Y, Llopis J, Tsien RY (2002) New biarsenical ligands and tetracysteine motifs for protein labeling in vitro and in vivo: synthesis and biological applications. J Am Chem Soc 124:6063–6076
Bruggisser J, Käser S, Mani J, Schneider A (2017) Biogenesis of a mitochondrial outer membrane protein in Trypanosoma brucei: targeting signal and dependence on a unique biogenesis factor. J Biol Chem 292:3400–3410
Connolly CN, Futter CE, Gibson A, Hopkins CR, Cutler DF (1994) Transport into and out of the Golgi complex studied by transfecting cells with cDNAs encoding horseradish peroxidase. J Cell Biol 127:641
De Mey J, Moeremans M, Geuens G, Nuydens R, De Brabander M (1981) High resolution light and electron microscopic localization of tubulin with the IGS (immuno gold staining) method. Cell Biol Int Rep 5:889–899
Denk W, Horstmann H (2004) Serial block-face scanning electron microscopy to reconstruct three-dimensional tissue nanostructure. PLoS Biol 2:e329
Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien RY, Ellisman MH (2002) Multicolor and electron microscopic imaging of connexin trafficking. Science 296:503
Giepmans BNG, Deerinck TJ, Smarr BL, Jones YZ, Ellisman MH (2005) Correlated light and electron microscopic imaging of multiple endogenous proteins using Quantum dots. Nat Methods 2:743–749
Grabenbauer M, Geerts WJC, Fernadez-Rodriguez J, Hoenger A, Koster AJ, Nilsson T (2005) Correlative microscopy and electron tomography of GFP through photooxidation. Nat Methods 2:857–862
Griparic L, van der Wel NN, Orozco IJ, Peters PJ, van der Bliek AM (2004) Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J Biol Chem 279:18792–18798
Hamer DH (1986) Metallothionein. Annu Rev Biochem 55:913–951
Harris KM, Perry E, Bourne J, Feinberg M, Ostroff L, Hurlburt J (2006) Uniform serial sectioning for transmission electron microscopy. J Neurosci 26:12101
Hart RG (1968) Electron microscopy of unstained biological material: the polytropic montage. Science 159:1464
Hopkins C, Gibson A, Stinchcombe J, Futter C (2000) Chimeric molecules employing horseradish peroxidase as reporter enzyme for protein localization in the electron microscope. In: Jeremy Thorner SDE, John NA (eds) Methods in enzymology, vol 327. Academic Press, New York, pp 35–45
Horstmann H, Körber C, Sätzler K, Aydin D, Kuner T (2012) Serial section scanning electron microscopy (S3EM) on silicon wafers for ultra-structural volume imaging of cells and tissues. PLoS ONE 7:e35172
Huang P, Yu T, Yoon Y (2007) Mitochondrial clustering induced by overexpression of the mitochondrial fusion protein Mfn2 causes mitochondrial dysfunction and cell death. Eur J Cell Biol 86:289–302
James DI, Parone PA, Mattenberger Y, Martinou J-C (2003) hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem 278:36373–36379
Kagi JH, Schaffer A (1988) Biochemistry of metallothionein. Biochemistry 27:8509–8515
Knott G, Marchman H, Wall D, Lich B (2008) Serial section scanning electron microscopy of adult brain tissue using focused ion beam milling. J Neurosci 28:2959
Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC (2004) Structural basis of mitochondrial tethering by mitofusin complexes. Science 305:858
Koster AJ, Chen H, Sedat JW, Agard DA (1992) Automated microscopy for electron tomography. Ultramicroscopy 46:207–227
Ladinsky MS, Mastronarde DN, McIntosh JR, Howell KE, Staehelin LA (1999) Golgi structure in three dimensions: functional insights from the normal rat kidney cell. J Cell Biol 144:1135
Lam SS, Martell JD, Kamer KJ, Deerinck TJ, Ellisman MH, Mootha VK, Ting AY (2015) Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat Methods 12:51–54
Li J, Wang Y, Chiu S-L, Cline HT (2010) Membrane targeted horseradish peroxidase as a marker for correlative fluorescence and electron microscopy studies. Front Neural Circuits 4:6
Li W, Ding W, Ji G, Wang L, Zhang J, Sun F (2016) Three-dimensional visualization of arsenic stimulated mouse liver sinusoidal by FIB-SEM approach. Protein Cell 7:227–232
Liu T, Yu R, Jin S-B, Han L, Lendahl U, Zhao J, Nistér M (2013) The mitochondrial elongation factors MIEF1 and MIEF2 exert partially distinct functions in mitochondrial dynamics. Exp Cell Res 319:2893–2904
Martell JD, Deerinck TJ, Sancak Y, Poulos TL, Mootha VK, Sosinsky GE, Ellisman MH, Ting AY (2012) Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat Biotechnol 30:1143–1148
McBride HM, Millar DG, Li JM, Shore GC (1992) A signal-anchor sequence selective for the mitochondrial outer membrane. J Cell Biol 119:1451
Meeusen S, McCaffery JM, Nunnari J (2004) Mitochondrial fusion intermediates revealed in vitro. Science 305:1747
Meißlitzer-Ruppitsch C, Vetterlein M, Stangl H, Maier S, Neumüller J, Freissmuth M, Pavelka M, Ellinger A (2008) Electron microscopic visualization of fluorescent signals in cellular compartments and organelles by means of DAB-photoconversion. Histochem Cell Biol 130:407–419
Mercogliano CP, DeRosier DJ (2007) Concatenated metallothionein as a clonable gold label for electron microscopy. J Struct Biol 160:70–82
Morphew MK, O’Toole ET, Page CL, Pagratis M, Meehl J, Giddings T, Gardner JM, Ackerson C, Jaspersen SL, Winey M, Hoenger A, McIntosh JR (2015) Metallothionein as a clonable tag for protein localization by electron microscopy of cells. J Microsc 260:20–29
Mozdy AD, McCaffery JM, Shaw JM (2000) Dnm1p Gtpase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J Cell Biol 151:367–380
Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K (2010) Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol 191:1141–1158
Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT (2011) MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep 12:565–573
Peddie CJ, Collinson LM (2014) Exploring the third dimension: volume electron microscopy comes of age. Micron 61:9–19
Rojo M, Legros F, Chateau D, Lombès A (2002) Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci 115:1663
Santel A, Fuller MT (2001) Control of mitochondrial morphology by a human mitofusin. J Cell Sci 114:867
Seligman AM, Karnovsky MJ, Wasserkrug HL, Hanker JS (1968) Nondroplet ultrastructural demonstration of cytochrome oxidase activity with a polymerizing osmiophilic reagent, diaminobenzidine (DAB). J Cell Biol 38:1
Shu X, Lev-Ram V, Deerinck TJ, Qi Y, Ramko EB, Davidson MW, Jin Y, Ellisman MH, Tsien RY (2011) A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS Biol 9:e1001041
Smirnova E, Griparic L, Shurland D-L, van der Bliek AM (2001) Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 12:2245–2256
Song Z, Chen H, Fiket M, Alexander C, Chan DC (2007) OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol 178:749
Su Y, Nykanen M, Jahn KA, Whan R, Cantrill L, Soon LL, Ratinac KR, Braet F (2010) Multi-dimensional correlative imaging of subcellular events: combining the strengths of light and electron microscopy. Biophys Rev 2:121–135
Waizenegger T, Stan T, Neupert W, Rapaport D (2003) Signal-anchor domains of proteins of the outer membrane of mitochondria: structural and functional characteristics. J Biol Chem 278:42064–42071
Yoon Y, Krueger EW, Oswald BJ, McNiven MA (2003) The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol 23:5409–5420
Youle RJ, Van Der Bliek AM (2012) Mitochondrial fission, fusion, and stress. Science 337:1062–1065
Zhao J, Liu T, Jin S, Wang X, Qu M, Uhlén P, Tomilin N, Shupliakov O, Lendahl U, Nistér M (2011) Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J 30:2762–2778
Acknowledgements
We would like to thank Prof. Quan Chen from Institute of Zoology, Chinese Academy of Sciences, for his kindness of giving the gene of Mfn2. We would like to thank Prof. Long Miao from Institute of Biophysics, Chinese Academy of Sciences, for his kindness of sharing the original plasmid of APEX. We are also grateful to Ping Shan and Ruigang Sui (F.S. lab) for their assistances. This work was supported by grants from the Strategic Priority Research Program of Chinese Academy of Sciences (XDB08030202) and the National Basic Research Program (“973 Program”) of Ministry of Science and Technology of China (2014CB910700). All the EM works were performed at Center for Biological Imaging (CBI, http://cbi.ibp.ac.cn), Institute of Biophysics, Chinese Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Yang Shi, Li Wang, Jianguo Zhang, Yujia Zhai, and Fei Sun declare that they have no conflict of interest.
Human and animal rights and informed consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Shi, Y., Wang, L., Zhang, J. et al. Determining the target protein localization in 3D using the combination of FIB-SEM and APEX2. Biophys Rep 3, 92–99 (2017). https://doi.org/10.1007/s41048-017-0043-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41048-017-0043-x