
Abstract
Noonan syndrome (NS) is a relatively common genetic syndrome with variable features including short stature, congenital heart disease, distinctive facial characteristics, skeletal anomalies, and varying degrees of developmental delay. NS is caused by gene mutations in a cellular signaling pathway that is essential for typical growth and development. Research in the past few decades has revealed that individuals with NS have highly variable neurocognitive and behavioral outcomes. To a certain extent, variability in cognitive functioning depends on the particular gene where a mutation is found; additionally, factors related to medical severity and environmental effects contribute substantially to outcomes. This article contains a systematic review of research on neuropsychological features of NS, including cognition, motor development, language, memory, attention, visual perception, adaptive behavior, social skills, and mental health concerns. Given the wide variety of possible cognitive and behavioral complications associated with NS, it is recommended that clinical neuropsychological evaluation of cognitive, adaptive, and psychological domains of functioning should be standard of care for all individuals with NS. Suggested considerations for assessment of individuals with NS are provided. The paper concludes with recommendations for educational and therapeutic interventions, as well as suggestions for future research directions.
Similar content being viewed by others

Introduction
Noonan syndrome (NS) is a genetic condition that was initially described by Dr. Jacqueline Noonan in 1963 (Noonan and Ehmke 1963). NS is equally common in males and females. The syndrome can either be inherited from an affected parent or can occur spontaneously due to a random gene mutation. Although there have not been any systematic epidemiological studies on NS, this condition has been estimated to occur in approximately 1:1,000 to 1:2,500 live births (Mendez and Opitz 1985). Clinically, NS is associated with craniofacial characteristics including widely spaced eyes, ptosis, low set ears, low posterior hairline, and webbed neck (Sharland et al. 1992). Cardiovascular abnormalities are seen in >80 % of individuals, with pulmonary valve stenosis, hypertrophic cardiomyopathy, and atrial septal defect occurring most frequently (Roberts et al. 2013). Individuals with NS are at increased risk for feeding and gastrointestinal problems, growth and endocrine disorders, delayed puberty, hematologic symptoms, chest deformity, scoliosis, and chronic pain, among other features (Romano et al. 2010; Smpokou et al. 2012). Hearing and visual concerns are also common (Qiu et al. 1998; Alfieri et al. 2008). Comprehensive reviews of medical features in NS were recently published by Romano et al. (2010) and Roberts et al. (2013). A consensus statement on clinical management guidelines is available online (Noonan Syndrome Guideline Development Group 2010).
A growing body of research reveals that neuropsychological impairments are quite common in NS (Wingbermühle et al. 2009). Indeed, for individuals who do not experience significant medical complications, concerns related to cognitive, social, or behavioral functioning could be the most significant component of having NS affecting their lives. Therefore, understanding how neuropsychological features impact patient outcomes and well-being is critical to providing quality health care for individuals with NS.
The purpose of the current paper is to provide an up-to-date review of the literature on neurocognitive and behavioral functioning in individuals with NS and to discuss the implications of these findings for clinical assessment practices. The paper begins with a brief overview of the molecular genetic basis of NS as well as a review of known neurological features, in order to provide the reader with a basic understanding of how biological mechanisms contribute to this unique disorder. Next, a comprehensive literature review is presented, covering each of the major domains of neuropsychological functioning. This review is followed by a section describing suggested considerations for neuropsychological assessment of individuals with NS. Recommendations for educational support and treatment of neuropsychological concerns are also offered. Although there have not yet been any clinical trials in NS targeting neurocognitive endpoints, there are a number of recommendations that can be formulated based on what is known about the medical and behavioral phenotype in NS and empirically supported interventions for individuals with similar conditions. Finally, proposals for future research directions are reviewed.
Molecular Genetic Basis of Noonan Syndrome: a Brief Overview
NS belongs to a family of related genetic syndromes that are referred to as the “RASopathies.” This term was derived because each of the RASopathy syndromes is caused by gene mutations that result in dysregulated activity of “Ras” proteins or related molecules. Ras proteins are important for normal functioning of cells throughout the body (Rauen 2013). These proteins operate within a cellular signaling pathway known as the Ras mitogen-activated protein kinase (MAPK) signaling pathway. This signal transduction pathway plays an essential role in embryonic and postnatal development. NS is caused by germline mutations in one of several genes that encode protein components or regulators within the RAS-MAPK pathway (Tartaglia et al. 2011; Aoki et al. 2013). Notably, gene mutations affecting other components of the RAS-MAPK pathway are known to cause several related genetic disorders, including neurofibromatosis type 1 (NF1), Legius syndrome, Costello syndrome, and cardiofaciocutaneous (CFC) syndrome. Due to a shared mechanism of dysregulation in the RAS-MAPK signaling pathway, these conditions have overlapping clinical features. The various genes that are associated with each of the RASopathies are listed in Table 1, along with the chromosome where each gene is located.
Although the different gene mutations that cause NS tend to result in similar sets of clinical features, a few specific variants have been associated with unique “Noonan-like” phenotypes. One variant causes a specific pattern of features that includes freckle-like skin markings (“lentigines”) and is referred to as Noonan syndrome with multiple lentigines (NSML; formerly known as LEOPARD syndrome). This disorder is allelic to NS and caused by specific mutations in the same genes, specifically in PTPN11 or RAF1 (Rauen 2013). Another variant is caused by mutations in the SHOC2 gene and results in a phenotype that has been described by the term Noonan-like syndrome with loose anagen hair (NS/LAG) (Cordeddu et al. 2009). The fact that different RASopathy genes can be associated with multiple disorders, or with a unique presentation of a disorder, underscores the complexity of the molecular genetic basis of NS. Indeed, the clinical presentation of NS and related disorders can vary widely across generations within a family (Bertola et al. 2004) and can also change considerably across an individual’s lifespan. These findings suggest that a great deal of the variability in how a person expresses a RASopathy syndrome depends on factors other than the individual’s specific genotype.
Neurological Features
Neurological conditions can occur in NS but are not a principal feature. Seizure disorders have been reported to occur in 10–13 % of affected individuals (Shaw et al. 2007). Multiple subtypes of seizures are possible, including generalized seizures, temporal lobe epilepsy, and febrile seizures (Sharland et al. 1992). Although craniofacial characteristics are associated with NS (e.g., widely spaced eyes, ptosis, triangular shaped face, low set ears, low hairline), the available research suggests that significant structural brain abnormalities are rare. Several case studies have reported an association between NS and Chiari I malformations. Chiari I malformations are a brain anomaly involving the inferior displacement and herniation of the cerebellar tonsils through the foramen magnum into the cervical canal. In studies of adults with NS, Chiari I malformation has been self-reported in 4–20 % of participants (Smpokou et al. 2012; Noonan 2005). However, reliable statistics from imaging data are not available to convincingly determine whether individuals with NS are at heightened risk for Chiari I anomalies or whether this association is coincidental (Keh et al. 2013). Hydrocephalus has also been described in NS, as well as a few isolated reports of structural cerebrovascular abnormalities, including cerebral arteriovenous malformations, cerebral cavernous malformations, aneurysms, and moyamoya disease (Zarate et al. 2013). For the most part, the data have been too limited to determine whether brain abnormalities can be linked to specific gene mutations in NS. As an exception, one study reported a distinct pattern of neuroanatomical abnormalities found in imaging studies of a few children with SHOC2 gene mutations, which included relative megalencephaly, mildly enlarged subarachnoid spaces, and small posterior fossa (Gripp et al. 2013). Overall, given the absence of obvious neuroanatomical findings in most individuals with NS, it appears unlikely that the neuropsychological impairments described below can be fully explained by major structural cerebral anomalies.
Materials and Methods
Literature Review
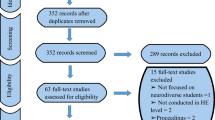
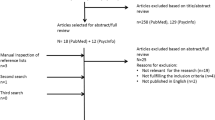
This comprehensive review encompasses all peer-reviewed empirical group studies from the past 25 years reporting on neuropsychological and behavioral functioning of individuals with Noonan syndrome. Studies were identified through searches within the PsycINFO and PubMed databases using the term Noonan syndrome combined with the following search terms: cognition, memory, visual, attention, motor, adaptive, behavior, emotion, social, autism, and quality of life. Full-text manuscripts were obtained for relevant abstracts and reviewed by the author. Reference sections of all articles were also reviewed to identify additional studies. Searches were limited to English language articles published between the years 1991 and 2015. Articles were excluded if they were not peer-reviewed group studies describing more than three patients. Table 2 includes the 19 publications that were identified which met the inclusion criteria. Although the majority of published studies on NS have examined neuropsychological functioning in children and adolescents, studies of adults with NS were also included to provide lifespan developmental context. Additionally, information from case study reports is mentioned within the review where appropriate to provide insights for which minimal data is otherwise available.
Intellectual Functioning
When large group comparisons are reported, intellectual abilities in children with Noonan syndrome tend to be mildly lowered as compared to unaffected children. The percentage of individuals with NS scoring in the IQ range associated with intellectual disability (IQ < 70) varies from 6 to 23 % across studies (van der Burgt et al. 1999; Pierpont et al. 2015), representing an increased risk relative to the general population. Despite this increased risk, the majority of people with NS have intellectual functioning within the average range (Pierpont et al. 2009; Wingbermühle et al. 2012b). It is notable that more recent studies report somewhat higher IQ estimates (Table 3). One possible explanation for this finding is that individuals with other RASopathy syndromes similar to NS could have been included in the earlier studies, thus contributing to lower overall IQ among the participants. As more comprehensive genetic testing for NS and other RASopathies has become available, better diagnostic precision among research samples has been possible. Regarding gender comparisons, larger studies have generally failed to find differences in intellectual functioning between boys and girls with NS (e.g., Pierpont et al. 2009).
Published studies do not suggest a clear pattern with regard to intellectual strengths and weaknesses in NS, although many individuals do show significant discrepancies across domains. Some studies have reported that children with NS have stronger verbal reasoning abilities relative to nonverbal reasoning abilities (Pierpont et al. 2009; van der Burgt et al. 1999), whereas other studies have found the opposite pattern (D. A. Lee et al. 2005) or no significant difference between these domains (Pierpont et al. 2015). Variation in the measures used or the age range studied (see Table 3) could potentially contribute to differences in the overall cognitive profile reported in various studies. Regardless of these measurement issues, it is clear that the cognitive phenotype is not consistent across individuals, highlighting the fact that relative strengths and weaknesses in intellectual functioning must be independently assessed for any given child.
All of the published studies involving children with NS have reported that affected individuals score lower than normative samples (or a sibling comparison group) on tests of intellectual ability. In contrast, there is some evidence to suggest that adults with NS may have less marked intellectual deficits. Wingbermühle et al. (2012a) reported no significant difference in full-scale IQ between a group of adult participants with NS and community control participants matched for age, gender, and education level. The authors noted that while the adults with NS in their cohort showed impaired performance compared with normative data, these norms did not adjust for levels of educational attainment and therefore may overestimate cognitive deficits in NS. The only intellectual subdomain in which a significant difference was found between the adults with NS, and the comparison group was in speed of information processing, which was significantly slower in the NS group. Slowed processing speed has been linked to a broad range of developmental disorders, including individuals with learning disabilities, attention problems, and mood/behavioral disorders (Willcutt et al. 2008). In both adults and children, speed of information processing is associated with maturation and structural organization of white matter tracts in the brain (Scantlebury et al. 2014). Processing speed could also be impacted by some medications which are taken by a minority of NS patients (e.g., seizure medication, chemotherapy drugs, pain relievers), although data related to historical or current medication use has not been reported alongside neurocognitive outcomes.
Theoretically, a large number of factors may contribute to the intellectual impairments that are seen in some individuals with NS. These include neurobiological risk factors arising from altered Ras signaling, as well as medical factors associated with the syndrome (e.g., congenital heart disease, perinatal risk factors, slowed growth and nutrition, poor energy/fatigue, hearing loss). In terms of medical predictors, van der Burgt et al. (1999) reported that children with a more severe physical expression of NS tended to have less favorable cognitive outcomes. In this study, children with a greater number of physical characteristics (based on a clinical severity rating involving facial features, heart disease, short stature, and chest wall abnormalities) had significantly lower verbal IQ and fluid reasoning as compared to those exhibiting fewer physical criteria. With respect to cardiac risk, Pierpont et al. (2009) did not find a significant association between heart disease severity ratings and verbal or nonverbal cognitive abilities in a relatively large sample of children with NS. While congenital heart disease is known to confer risk for neurocognitive deficits, intellectual impairments are most frequently seen for cyanotic conditions or those requiring open heart surgery in the neonatal period (Marino et al. 2012), which occur in a relatively small percentage of children with NS (Colquitt and Noonan 2014). For individuals with NS who have more severe cardiac conditions requiring surgery, no studies have examined whether specific operative or postoperative factors may give rise to differences in cognition or behavior. Additionally, no studies have directly examined whether nutritional or growth factors are associated with cognitive abilities in NS. Regarding sensory functions, one study reported that hearing loss in NS was associated with greater impairments on verbal reasoning tasks (Pierpont et al. 2009). Hearing loss is more common in children with NS than in the general population (Qiu et al. 1998), and many individuals experience frequent ear infections in childhood (Sharland et al. 1992).
Motor Development
Development of fine and gross motor skills can be significantly delayed in NS, especially during infancy and preschool years. It is likely that some degree of motor delay can be ascribed to health-related features of NS that affect physical development, which include hypotonia (weak muscle tone), failure to thrive, congenital heart disease, and skeletal anomalies (Tartaglia et al. 2011). Regarding developmental milestones, a large natural history study reported that the average age at which children with NS are able to sit unsupported is around 10 months, and average age for first walking is 21 months (Sharland et al. 1992). In a study involving caregiver report of adaptive functioning, children with NS ages 1–6 years were found to have greater delays in motor skills relative to communication and social skills (Pierpont et al. 2010b). Gross motor skills were more significantly delayed than fine motor skills.
Motor skills continue to be affected throughout childhood and adolescence. Lee et al. (2005) administered a motor and manual skills test battery to a set of school-aged children with NS and reported that 51 % of the sample scored in the range indicating presence of developmental coordination disorder. In another study of school-aged children, 75 % performed in the impaired range on a motor assessment battery which measured manual dexterity, ball skills, and balance (Alfieri et al. 2011a). Pierpont et al. (2009) reported that manual motor speed and dexterity was severely impaired (>2 SD below the mean) in 34 % and below average (>1 SD below the mean) in 72 % of preschool and school-aged children with NS who were assessed using the Purdue Pegboard test (Tiffen 1968). In this study, manual dexterity was found to be significantly correlated with both verbal and nonverbal intellectual functioning. Taken together, these studies suggest that coordination and fine motor problems may be one of the most common neuropsychological deficits among individuals with NS and that performance on motor and cognitive tasks is interrelated.
Visual Processing
Problems with visual perception and processing appear to be common in NS, although few studies have examined these abilities independent of motor ability. NS is associated with a number of ocular anomalies. These include external features (e.g., ptosis, widely spaced eyes, downward-slanting palpebral fissures) as well as conditions like strabismus and nystagmus (Marin et al. 2012). Refractory errors affect about 2/3 of individuals with NS (Lee et al. 1992). Alfieri et al. (2008) administered examinations of visual function to 24 individuals with NS or NSML and reported abnormal results on 83 % of the tests involving ocular movements, acuity, and stereopsis. Another study assessed school-aged children with NS using computerized tests of visual processing abilities involving form and motion coherence (Alfieri et al. 2011a). Children with NS performed significantly worse on tests of form coherence (39 % impaired) relative to motion coherence (11 % impaired). With regard to the neural basis of this finding, the authors suggested that children with NS may have greater difficulties with ventral stream processing (“what” pathway used for object identification) compared with dorsal stream processing (“where” pathway used for spatial processing).
A few studies have examined visual–motor integration skills in children with NS. One study reported that 33 % of children with NS or NSML had significant impairment, and 48 % had minor impairment on a standard visual–motor integration task requiring the examinee to copy a set of increasingly complex geometric shapes (Alfieri et al. 2008). In this study, it was not possible to determine whether the impaired performance of some children was related to difficulties with visual perception, motor impairment, or a combination of these skills. In adults with NS, visual–motor integration was assessed using the copy trial of the Rey-Osterreith Complex Figure Test (Rey 1941). Adults with NS performed equivalently on this task to adults in a comparison group (Wingbermühle et al. 2012b).
Speech and Language
In terms of speech and language milestones in young children with NS, average age at first word has been reported to be around 15 months (Pierpont et al. 2010b), with simple two-word phrases emerging around 31–32 months (Sharland et al. 1992; Sarimski 2000). Significant feeding difficulties affect about 75 % of children with NS (e.g., poor suck/swallow, gagging, reflux, recurrent vomiting, food refusal, and intestinal dysmotility) and often generate referrals to a speech-language pathologist or feeding specialist (Shah et al. 1999; Romano et al. 2010). Parent reports of adaptive functioning indicate that expressive language skills tend to develop more slowly than receptive language skills in infants and preschool children with NS (Pierpont et al. 2010b). A majority of children with NS (70 %) receive speech/language therapy, and about half (48 %) use simplified sign language as a means of communication or as a supplement to speech at some point in development (Pierpont et al. 2010a).
Communication skills of school-aged individuals with NS were investigated by Pierpont et al. (2010a), who completed extensive speech and language evaluations in a large group of children and adolescents. Performance on standardized language assessments was lower than expected based on normative data, with 30 % of participants showing evidence of language impairment (i.e., scoring <10th percentile in receptive and/or expressive language ability). Articulation impairment was evident among 20 % of the children and was found to be modestly correlated with language ability. This study identified several cognitive factors that were associated with language development in NS. Measures of nonverbal intellectual functioning and phonological memory emerged as the strongest predictors of language competence. Additionally, children who passed an audiological screening also scored higher on tests of language and articulation than those who had abnormal audiological test results, suggesting that hearing loss or poor auditory processing may be important risk factors affecting speech-language development in children with NS.
Problems with language development appear to be associated with delayed development of academic skills in children with NS. Reading and spelling skills have been reported to be strongly correlated with both language ability, phonological memory, and nonverbal cognitive ability in this population (Pierpont et al. 2010a). Preliminary evidence also suggests that social aspects of language may be affected in NS. In the study by Pierpont et al. (2010a), 60 % of children with NS were rated as having probable pragmatic language impairments based on parent ratings on a standardized measure. Notably, male participants were significantly more likely to have pragmatic language deficits than female participants. Studies employing direct assessment of pragmatic language and social communication in individuals with NS are greatly needed.
Memory
Memory formation and consolidation has become an area of particular interest in the field of RASopathies research. Molecules in the RAS-MAPK pathway are known to be essential for regulating brain plasticity, including processes necessary for memory stability (Davis and Laroche 2006). Animal models have demonstrated that mutations mimicking those in NS result in deficits in long-term memory formation in the hippocampus or analogous regions and are associated with learning deficits (Pagani et al. 2009; Lee et al. 2014). As a result, there has been increased attention in trying to understand whether learning deficits may be reversed or attenuated using pharmacological treatments or alternative learning methods that capitalize on our understanding of the mechanisms interfering with memory encoding and consolidation.
A handful of studies have examined specific learning and memory processes in people with NS. Alfieri et al. (2011b) reported that 50 % of a cohort of children with genetically confirmed NS demonstrated impaired performance on a verbal free recall task, whereas only 5–20 % demonstrated impairments on visual and spatial recognition memory tasks. Although results of this study suggest that some aspects of memory may be disproportionately affected in NS, it is unclear from this data whether deficits on the verbal task were due to the retrieval (i.e., free recall) demands or the verbal nature of the task. In a study by Pierpont et al. (2013), children with NS were administered tests of verbal, visual, and working memory from a standardized memory battery (WRAML-2; Sheslow and Adams 2003). Results indicated a relative strength on verbal memory tasks compared with visual memory and working memory tasks. Furthermore, within the verbal tasks, a significant advantage was seen in delayed recognition memory relative to delayed recall memory. These results are consistent with the finding from Alfieri et al. (2011b) that verbal free recall tasks that depend heavily on prefrontal–hippocampal networks are more challenging for children with NS than recognition tasks that rely on more distributed temporal cortical regions. Interestingly, Wingbermühle et al. (2012b) did not find significant deficits among adults with NS as compared to a comparison group with respect to verbal delayed recall on a list-learning task or delayed visual recall as measured by the Rey-Osterreith Complex Figure Test (Rey 1941). This finding suggests that memory problems may not be evident in adulthood, at least not relative to individuals with equivalent education levels. It is also possible that adults with NS may be more apt to use compensatory strategies to overcome learning and memory deficits.
Attention and Executive Functioning
Emerging research suggests that problems with attention and related functions are likely to be one of the most common neuropsychological impairments in children with NS, as has been found to be the case in other RASopathies such as NF1 (e.g., Isenberg et al. 2012; Payne et al. 2011; Galasso et al. 2014). Earlier studies using parent report measures such as the Child Behavior Checklist have noted children with NS showed greater difficulties related to attention and hyperactivity relative to comparison groups or normative data, although statistically significant differences were not always reported (van der Burgt et al. 1999; Wood et al. 1995; D. A. Lee et al. 2005; Sarimski 2000; Alfieri et al. 2014). More recently, Pierpont et al. (2015) compared a cohort of children with NS with a sibling comparison group on a range of measures examining attention skills. Relative to their unaffected siblings, children with Noonan syndrome had higher rates of past ADHD diagnosis (31 %). Additionally, parent ratings of ADHD symptoms were significantly higher in the children with NS when using both DSM criteria-based and broadband behavior scales. On direct behavioral measures assessing auditory attention, sustained attention, and response inhibition, children with NS showed significant impairments compared with unaffected siblings on all three measures. Attention regulation skills were more strongly correlated with intellectual test performance (WASI Full Scale IQ) in the Noonan syndrome group than the comparison group. This finding suggests that lower IQ may be a risk factor for attention difficulties in children with NS. Alternatively, it is also possible that ADHD symptoms could interfere with the ability of some children with NS to learn and retain information and/or to demonstrate their cognitive capacities in educational or testing environments.
A high rate of ADHD symptoms in NS is suggestive of possible neurodevelopmental differences in frontal lobe–subcortical circuitry, a conceptualization which is supported by studies of other RASopathies. Some research has indicated that Ras proteins (which are abnormally activated in these syndromes) may regulate interneuronal activity in prefrontal and striatal networks (Shilyansky et al. 2010a). These regions of the brain are known to be critical for a number of higher-order cognitive processes such as working memory, response inhibition, and cognitive flexibility (Powell and Voeller 2004). Working memory abilities are known to be an area of weakness for children with other RASopathies, most notably NF1 (Rowbotham et al. 2009; Shilyansky et al. 2010a). In line with this finding, Pierpont et al. (2013) reported that children with NS had weaker performance on a test assessing auditory and visual working memory than normative data, with 34 % showing significant impairment. In contrast, one study reported that adults with NS did not exhibit significant deficits on auditory working memory tasks (e.g., digit span) or other executive functioning tasks (e.g., Tower of London, Stroop Color Word Interference) relative to a comparison group (Wingbermühle et al. 2012b).
Results from questionnaire measures reveal that individuals with NS show functional deficits with everyday tasks that depend on executive functioning. Parent ratings indicate that children and adolescents with NS show markedly worse performance on tasks requiring working memory (e.g., forgetting steps within a task, having trouble concentrating on chores and activities) and self-monitoring (e.g., being unaware of how a behavior affects others, not checking work for mistakes, having a poor understanding one’s own strengths/weaknesses) compared with unaffected children (Pierpont et al. 2015). Similarly, adults with NS have reported significantly higher rates of subjective executive functioning problems than control participants when assessed using self-report ratings (Wingbermühle et al. 2012b). Additional research is needed to determine whether and how functioning and maturation of the frontal lobes and connected brain regions that underlie higher-order cognitive organization may differ in individuals with NS compared with the general population.
Social Functioning
Aside from attention problems, another area in which clinically significant difficulties are consistently found on standardized behavioral checklists is social skills competence (Pierpont et al. 2015; Alfieri et al. 2014; Wood et al. 1995; van der Burgt et al. 1999). Individuals with NS have been described as having tendencies toward social immaturity, reduced interaction with peers, and diminished insight in social situations (Wingbermühle et al. 2009; Sarimski 2000), although many of these characteristics have not been examined in depth. With respect to social cognitive processing, no studies have directly investigated whether children with NS have impairments on behavioral tasks assessing emotion perception, social attribution of meaning, or other aspects of social cognition. One study examined social affective processing in adults with NS. Wingbermühle et al. (2012a) reported that adults with NS performed more poorly than unaffected individuals on a task assessing perception and labeling of emotional facial expressions, although the effect size was small and no differences were found in the perception of any specific emotions. On questionnaire measures, adults with NS in this study also self-reported greater problems in processing emotional states. Follow-up analyses indicated that differences between adults with NS and controls were driven primarily by reported differences in ability to identify and communicate about emotional states (i.e., “alexithymia”) rather than by differences in the conscious experience of emotion-based arousal. With regard to the ability to take attribute mental states to self and others and engage in perspective-taking (“theory of mind”), research has shown no differences between adults with NS and unaffected adults (Wingbermühle et al. 2012a). Taken together, these findings suggest that mild or moderate impairments in social–emotional processing or communication may occur in NS, but the cognitive basis of these difficulties is as of yet unclear.
It has been suggested that one factor that could potentially impact social development and relatedness of individuals with NS is the experience of teasing or bullying (Verhoeven et al. 2008). Noonan (2005) surveyed a set of adult patients with NS, many of whom indicated that they were often teased as children because they had short stature or looked different from their peers. Unfortunately, the prevalence and severity of peer victimization among children with NS has not yet been systematically assessed. Possible effects of repeated peer victimization on psychological adjustment include depressed mood, interpersonal difficulties (e.g., social anxiety, social skills deficits, and loneliness), and academic problems (Storch and Ledley 2005). In individuals with NS, reactions to teasing/bullying could manifest as reduced comfort in social situations rather than overt social avoidance. Indeed, Wingbermühle et al. (2012a) found that adults with NS reported higher levels of distress related to interpersonal interactions than unaffected adults.
Autism Spectrum Disorder
At present, there is some evidence suggesting that children with NS and Noonan-like conditions are at heightened risk for autism spectrum disorder (ASD) relative to the general population, although the available research has relied on screening measures rather than comprehensive diagnostic assessments. One study by Alfieri et al. (2014) analyzed data from standardized parent report checklist measures, including the Social Communication Questionnaire (SCQ; Rutter et al. 2003) and the Modified Checklist for Autism in Toddlers (M-CHAT; Robins et al. 2001). An elevated level of ASD symptoms (relative to a conventional cutoff score maximizing sensitivity and specificity to ASD) was reported among 12 % of children with NS and 25 % of children with NSML. When evaluated clinically using DSM-IV-TR (American Psychiatric Association 2000) criteria, none of these children were found to meet diagnostic criteria for ASD. In another study by Adviento et al. (2014), 21 % of participants with NS showed elevated ASD symptoms on the SCQ screening measure, which was significantly different than the rate of elevated scores found in a sibling comparison group (0 %). Scores on the Social Responsiveness Scale (SRS; Constantino and Gruber 2005), another screening measure of ASD symptoms, were also significantly higher in the NS group than the sibling group. In-depth diagnostic testing using gold standard assessment of ASD was completed for one participant with NS in this study, who was not found to meet ASD criteria. Taken together, results of these studies suggest that although characteristics of ASD may be relatively common in NS, the vast majority of children will not meet full diagnostic criteria for the condition. Further research is needed to determine whether the heightened prevalence of autistic features in NS arises primarily from the intellectual, communication, and sensory impairments that are common in NS or whether these behaviors are associated with more pervasive deficits in social reciprocity or social cognition.
Psychopathology/Mental Health
While long-term follow-up studies of adults suggest the possibility that mood and anxiety disorders may be frequently comorbid with NS, this observation has not yet been verified with controlled studies. Noonan (2005) reported that 23 % of adults with NS self-reported having been diagnosed with depression and taking antidepressant medication. In another study, 49 % of adult patients self-reported having been diagnosed and treated for depression and/or anxiety (Smpokou et al. 2012). In contrast to these findings, studies using standardized symptoms checklists have not found a significant difference in anxiety and depression symptoms in adults with NS compared with unaffected adults (Wingbermühle et al. 2012a). Similarly, most studies have found no differences in parent-reported levels of depression/anxiety symptoms between children with NS compared with typically developing children (Pierpont et al. 2015; Wood et al. 1995; van der Burgt et al. 1999), although Alfieri et al. (2014) reported a 32 % rate of clinically elevated depression/anxiety symptoms in their sample of children with NS. In terms of self-concept, Lee et al. (2005) administered the Piers-Harris Children’s Self-Concept Scale (Piers and Harris 1989) to assess self-esteem in 30 children with NS. The mean score and standard deviation for children with NS on this assessment were not significantly different from those of a standardized population. Taken together, these results suggest that if mood disorders do indeed occur more commonly in NS, the symptoms are most likely to arise sometime during or after adolescence and may not be clearly evident in self-report measures of emotional symptomatology. The possibility of under-reporting of emotional symptoms should be explored, given that individuals with NS have been described as having a tendency toward offering socially desirable responses (Verhoeven et al. 2008).
A handful of adult case reports have reported co-occurrence of NS and other psychiatric diagnoses including alcohol abuse, anorexia nervosa, bipolar disorder panic disorder, obsessive-compulsive disorder, and schizophrenia (Arvaniti et al. 2014; Noonan 2005; Wingbermühle et al. 2009). Nevertheless, given the dearth of these types of published reports, there is no clear evidence to suggest that serious mental illness is more common in NS than in the general population. In terms of behavioral symptoms, parents of children with NS reported more significant attention-seeking behaviors compared to the general population, which may result in mildly higher rates of parenting stress related to these behaviors (Sarimski 2000). One study reported a 34 % rate of clinically significant externalizing symptoms in a sample of children with NS (Alfieri et al. 2014), but the threshold for significance was not described and scores were not compared to a control sample. In contrast, other studies have not found that aggressive behaviors and other externalizing symptoms are observed more frequently in individuals with NS than in typically developing individuals (Pierpont et al. 2015; Wingbermühle et al. 2009).
Adaptive Behavior
Adaptive behavior refers to the set of conceptual, social, and practical skills that are necessary for functioning in everyday life (Luckasson et al. 2002). Impairments in adaptive functioning can arise from a variety of sources, such as cognitive deficits, physical limitations, illness, emotional/behavioral dysregulation, or interpersonal problems. Pierpont et al. (2010a) provided data from a cohort of individuals with molecular confirmation of NS whose parents completed caregiver report measures of adaptive behavior. Adaptive behavior ratings of individuals with NS were significantly lower than the normative population, with a group mean in the low average range. Adaptive behavior was roughly commensurate with the expected range based on studies of intellectual functioning. There were no differences in ratings of boys and girls with NS. Scores of younger children with NS (ages 1–6) revealed relative weaknesses in motor skills and daily living skills (e.g., self-care, community participation) as compared to social and communication skills. This finding may reflect the fact that many children with NS experience hypotonia, feeding problems and delayed physical development in early life. By school age and adolescence, children with NS do not show significant discrepancies across the different domains of adaptive functioning. Parent ratings in this study indicated that expressive language skills, which initially lag behind receptive skills during infancy and preschool years, tend to be roughly equivalent to receptive language skills by school age.
In terms of risk factors for adaptive impairments, gestational age at birth has been found to be a significant predictor of adaptive functioning, such that children with NS and CFC syndrome who were born preterm are at a higher risk for adaptive delays than those born full-term (Pierpont et al. 2010a). This finding is consistent with a large research literature showing that infants born prematurely have less favorable long-term educational and social outcomes (e.g., Moster et al. 2008; Lindstrom et al. 2007). Additionally, individuals with NS whose parents have higher education levels are at reduced risk for cognitive and adaptive difficulties (Pierpont et al. 2010a; Pierpont et al. 2009). Thus, variables related to parenting and the home environment may contribute significantly to development of important functional skills.
Quality of Life
Two studies have examined quality of life in individuals with Noonan syndrome. Both studies analyzed self-report questionnaire completed by adult European participants. In a small Dutch cohort of ten adults with NS, an average to high-average level of satisfaction was found in terms of quality of life across all domains (leisure and social participation, health, family relations, and fulfillment of life goals) (Verhoeven et al. 2008). A second study reported follow-up information about 45 adults with NS who had previously been recruited at a medical institution in Germany 25 years earlier (Binder et al. 2012). Consistent with the Dutch study, no significant difference was found between responses of individuals with NS and an age-matched healthy reference cohort on a 36-item quality of life survey. However, results of this study indicated that individuals with NS had significantly lower levels of education and graduation achievements than the general population and lived more frequently without a partner. Mortality rates were also higher among individuals with NS than the general population. Results of these studies indicate that although health, educational, and psychosocial (partnership and parenting) outcomes may be affected by NS, perceived quality of life does not differ markedly from reference populations. This finding suggests that by adulthood, many individuals show good adaptation to the experience of having NS.
Relationship Between Genotype and Neurocognitive Outcomes
When the RASopathies are considered collectively, there are fairly robust genotype–phenotype correlations with respect to neurocognitive functioning. A number of studies have shown that mutations in genes acting more downstream in the RAS-MAPK pathway (e.g., BRAF, MEK 1, MEK2, HRAS), which typically cause CFC syndrome and Costello syndrome, tend to be associated with greater impairments in neurological, intellectual, and adaptive functioning than mutations in genes encoding components or regulators that act more upstream in the pathway (e.g., NF1, PTPN11, SOS1, RAF1), which cause NF1 and NS (Cesarini et al. 2009; Pierpont et al. 2010b; Yoon et al. 2007). Individuals with gene mutations acting downstream in the pathway (especially those diagnosed with CFC syndrome) also tend to have a higher prevalence of autistic-like traits (Adviento et al. 2014; Alfieri et al. 2014).
When considering only individuals diagnosed with NS, the relationship between genotype and neurocognitive functioning tends to be more subtle. The largest amount of data is available for individuals with mutations in the PTPN11 gene. PTPN11 mutations are the most common cause of NS and account for approximately 50 % of cases (Tartaglia et al. 2001). Individuals with PTPN11 mutations have a wide spectrum of neuropsychological outcomes, with large variation evident among those with mutations within the same exon and even among those with the exact same sequence variation (Pierpont et al. 2009). Nevertheless, there is some evidence that mutations at certain positions could be protective with regard to neurocognitive outcomes. In an analysis of a large cohort of individuals found to have PTPN11 mutations, Tartaglia et al. (2002) reported that all 17 individuals with the p.N308D mutation were found to attend regular education classrooms, suggesting that neurocognitive disabilities may be milder in this set of individuals. In line with this, Pierpont et al. (2009) reported that six individuals with p.N308D and p.N308S mutations performed within normal limits in IQ testing. This finding suggests that these sequence variations are not frequently associated with cognitive disabilities. Additional research suggests that PTPN11 mutations associated with the NSML phenotype are also often associated with normal intellectual functioning (Sarkozy et al. 2008), although direct testing results have been published for only a few patients with this clinical diagnosis (Alfieri et al. 2014).
Mutations in the SOS1 gene cause approximately 15 % of cases of NS (Roberts et al. 2007). Most research suggests that individuals with mutations in the SOS1 gene have relatively favorable outcomes with regard to cognitive development. In one study, intellectual functioning of children with SOS1 mutations was found to be significantly higher than those with PTPN11 mutations, as well as those with unknown mutations (Pierpont et al. 2009), although the sample size of the SOS1 mutation group was relatively small. This finding is consistent with other reports suggesting that, in the absence of complicating neurological features such as seizures, most people with SOS1 mutations have intellect within the average range and attend regular education classrooms (Tartaglia et al. 2007; Cesarini et al. 2009). In general, behavioral and adaptive functioning of children with SOS1 mutations tends to be consistent with those with PTPN11 mutations and those of other children with a clinical diagnosis of NS (Pierpont et al. 2010b; Pierpont et al. 2015; Alfieri et al. 2014).
Rarer gene mutations, including those in NRAS, KRAS, RIT1, RAF1, SCHOC2, and CBL, account for relatively few cases of NS (Aoki et al. 2013; Roberts et al. 2013). Studies reporting on individuals with RAF1 mutations typically find that intellectual and adaptive functioning is commensurate with individuals with other NS gene mutations (Cesarini et al. 2009; Pierpont et al. 2010b; Alfieri et al. 2014). Neuropsychological functioning in individuals with NRAS, CBL, and RIT1 mutations is largely unknown. There is some evidence to suggest that individuals with KRAS and SHOC2 mutations may experience greater severity of neurological deficits and developmental delay compared to other children with a diagnosis of NS, with intellectual disability being relatively common (Lee et al. 2011; Cesarini et al. 2009; Alfieri et al. 2014; Gripp et al. 2013).
The wide spectrum of developmental outcomes in the RASopathies, along with significant clinical overlap in the phenotype, results in some ambiguity in clinical classification of individuals, particularly in situations where neurological involvement is one of the key distinguishing features. For example, individuals with KRAS mutations have been described as having an intermediate or ambiguous clinical phenotype that has features of both NS and CFC syndrome (Schubbert et al. 2006; Nystrom et al. 2008). Individuals with this mutation can be diagnosed with either one of these conditions depending on their specific clinical presentation. Interestingly, one study found that two individuals with KRAS mutations each scored between the mean of the CFC-confirmed and NS-confirmed groups in terms of overall scores on adaptive functioning measures, suggesting that these individuals may also exhibit a somewhat intermediate profile in terms of neurocognitive functioning (Pierpont et al. 2010b). On a related note, some studies have found that, occasionally, mutations in BRAF and MEK1 can result in a phenotype more similar to NS or NSML than to typical CFC (Nystrom et al. 2008; Nishi et al. 2015; Sarkozy et al. 2009). This finding has resulted in some debate regarding whether these individuals can be diagnosed as having a Noonan-like syndrome or whether they simply exhibit a milder phenotype of CFC (Neri et al. 2008). Regardless of this classification issue, there are data to indicate that the spectrum of BRAF and MEK1 mutations includes individuals with unimpaired intellectual functioning, despite evidence that the vast majority of children with these mutations present with intellectual disability (Allanson et al. 2011; Koudova et al. 2009; Pierpont et al. 2009). Additional research is needed to determine whether particular sequence variants in these genes are reliably protective with respect to neurocognitive outcomes.
Discussion
The above review of cognitive and behavioral characteristics associated with Noonan syndrome and Noonan-like phenotypes suggests a number of general conclusions. First, delayed developmental milestones are extremely common in NS during the infancy and toddler years, especially with regard to gross motor development and expressive language. While more severe delays in language or motor skills appear to place a child with NS at risk for less favorable neurocognitive outcomes, it is important to note that by school age, overall intellectual functioning measures within normal limits in the majority of children. During later childhood and adolescence, language abilities tend to be generally commensurate with nonverbal cognitive functioning. Indeed, intellectual abilities tend to track along fairly closely with a variety of other neurocognitive domains, including attention, motor, and communication skills. This finding suggests that risk factors for neuropsychological impairments in NS often have somewhat generalized (global) effects on brain development. For children with average IQ, deficits in other domains of neurocognitive functioning, when present, can be relatively subtle.
Although there are no published longitudinal studies of neuropsychological functions in NS, the available data seem to suggest that adults with NS have generally milder cognitive deficits as compared to children, particularly with regard to domains such as attention, executive functioning, and memory. Several possible factors could contribute to this finding. First, by the time they enter adulthood, individuals with NS may develop the ability to compensate for some of their neurocognitive challenges. Secondly, Wingbermühle et al. (2012b) reported that although adults with NS demonstrated impaired performance relative to normative data, these deficits were less prominent when scores were compared to a control group matched on age and education level. This finding suggests a “schooling effect,” whereby cognitive deficits in NS may be overestimated due to differences in educational background. In light of this finding, it will be important to consider factors that may contribute to the reported discrepancies in educational attainment in this population. For example, it could be that delayed maturation of certain regions of the brain (e.g., the frontal lobes) could account for the apparent disparities in psychosocial outcomes (e.g., educational attainment, quality/maintenance of interpersonal relationships). Unlike performance on a cognitive test, these outcomes often depend on consistent functioning/achievement throughout the course of a person’s life. In line with this, there is some evidence to suggest that while cognitive functioning seems to remain stable or improve in NS throughout development, affected children do not always maintain pace with their peers with regard to their adaptive functioning. Cross-sectional data from parent report measures indicate the deficits in independent living skills (relative to peers) may be more prominent in older children and adolescents than in younger children (Pierpont 2010b).
In the last few years, greater information has become available regarding the relationship between genotype and neuropsychological development. Results of this review suggest that diverse neurocognitive outcomes are possible with all gene mutations associated with NS. For every gene associated with NS, there have been reports of individuals with intellectual functioning in the average range, as well as individuals with significant intellectual disabilities. Importantly, cognitive functioning can measure within the average range even among children with mutations typically associated a more neurologically involved phenotype (e.g., KRAS, SHOC2) or children with a Noonan-like phenotype who have mutations in genes acting more downstream in the RAS-MAPK pathway that are classically associated with CFC syndrome (e.g., BRAF, MEK1). As a result, gene mutation can never be considered a definite prognostic indicator. Nevertheless, for the majority of individuals with mutations in genes most commonly associated with NS (e.g., PTPN11, SOS1, RAF1), overall cognitive functioning is within normal limits. For these children, concerns related to other aspects of learning or behavior tend to be predominant. Additionally, some research suggests that within a given gene, specific sequence variants could be protective with regard to neuropsychological outcomes. Unfortunately, this research is often hampered by small numbers of individuals in a given study with the same mutation. Greater clarity with regard to genetic risk factors will undoubtedly emerge as molecular testing is more widely available, and these results are reported in conjunction with neuropsychological research.
A key finding emerging from empirical research is that regardless of their gene mutation, children with NS are at heightened risk for difficulties with attention regulation and reduced cognitive organization/flexibility. As is the case with many other neurodevelopmental and genetic disorders (Lo-Castro et al. 2011), ADHD symptoms appear to be one of the most common behavioral features in children with NS. Additionally, while it is unknown how prevalent “classic” learning disabilities are in NS (e.g., reading or mathematics disorders), it is evident that difficulties related learning and memory can commonly occur in NS. Memory difficulties appear to be particularly evident with regard to working memory, as well as in memory tasks requiring free recall of stored information. At present, it is unclear whether the learning problems that many children with NS experience in the classroom are attributable to one specific underlying problem or to multiple additive factors.
As stated earlier, it is unlikely that neuropsychological impairments can be fully explained by major structural brain anomalies. Since the discovery of genetic mutations which underlie NS and other RASopathies, there has been greater attention to understanding the potential mechanisms whereby altered Ras signaling may impact brain development or functioning in more subtle ways. Some of the most promising work in terms of understanding how altered gene expression may give rise to neurodevelopmental differences has come from animal models. For example, these models have shown that altered activation of SHP-2 (the PTPN11 gene product) can interfere with neural cell-fate decisions (Gauthier et al. 2007). Further, it has become increasingly clear that molecules in the RAS-MAPK pathway are essential for normal neurophysiologic functioning within multiple brain circuits (Samuels et al. 2009), including those involved in learning and memory (Costa et al. 2001; Pagani et al. 2009). Lee et al. (2014) recently demonstrated that mice expressing either of two NS-associated mutations in PTPN11 show hippocampal-dependent impairments in spatial learning tasks as well as deficits in hippocampal long-term potentiation. Other studies have reported that dysregulation of Ras can result in differences in maturation or functioning of frontal–subcortical circuitry involved in attention and cognitive control (Shilyansky et al. 2010a). In a study of humans, Mainberger et al. (2013) reported that, relative to a healthy comparison group, individuals with NS show reduced motor cortex plasticity (measured by changes in motor evoked potential amplitude) in response to paired peripheral electrical and transcranial magnetic stimulation. These findings suggest potential neural targets for intervention which, when combined with a clear understanding of how neuropsychological deficits impact daily lives of individuals with NS, could lead to major benefits for individuals with this syndrome.
Ultimately, it is likely that biochemical changes (e.g., dysregulated Ras signaling) can affect brain development differently in various individuals and that the interplay of cellular and molecular abnormalities with medical factors is what gives rise to the range of possible neurobehavioral concerns that have been described in this review. Indeed, research on NF1, a RASopathy that has a similarly wide range of clinical features as NS, supports the view there may be no single molecular abnormality that explains the neurocognitive effects seen in these genetic syndromes. It has been recently speculated that multiple developmental and physiological processes contribute to the specific collection of cognitive and behavioral deficits observed in any given child with NF1 (Diggs-Andrews and Gutmann 2013). As such, from an intervention perspective, it may be critical to adopt a personalized medicine approach, which utilizes targeted therapies specific to the biochemical and neurochemical abnormalities that arise in each individual. The identification of how each neuropsychological profile maps on to these neurobiological mechanisms poses an exciting challenge for future research on NS and other RASopathies.
Considerations for Clinical Neuropsychological Assessment of Individuals with NS
Given the vast array of possible neurological and neuropsychological issues related to NS, comprehensive assessment of each individual’s unique capacities will be essential to educational and intervention planning. Therefore, it is recommended that neuropsychological evaluation of cognitive, adaptive, and psychological domains of functioning should be standard of care for all individuals with NS. Pediatric neuropsychologists working in a medical setting (e.g., children’s hospitals, academic health centers, medical clinics) may be especially well-equipped to evaluate individuals with NS, given the complexity of the medical issues that can accompany this diagnosis. These assessments can be completed at key intervals throughout the lifespan, particularly when concerns arise for parents, teachers, medical providers, or individuals with NS themselves. In infancy or early childhood, assessments may focus primarily on identifying need for rehabilitation therapies. By middle childhood, neuropsychological evaluation may be very useful to inform educational services, classroom accommodations, or behavioral therapies. During adolescence or adulthood, assessments may focus on various targets including academic services in high school or college, transition or employment needs, and social or mental health supports.
When assessing young children with NS, neuropsychologists should be aware of the potential impact that weak oral motor or fine motor skills may have on intellectual assessments. For example, Pierpont et al. (2009) reported that the most challenging subtest for children with NS on the Differential Ability Scales (Elliott 1990) was the Recall of Designs subtest; this subtest relies more heavily on fine motor skills. Therefore, selection of at least some measures that are relatively independent of motor skills is suggested in order to attain the most accurate results. Independent assessment of finger and hand mobility may shed light on the potential contribution of these issues to test performance and daily academic and adaptive functioning. Additionally, for all individuals with NS, sensory concerns (e.g., vision or hearing limitations) must also be taken into account when selecting appropriate cognitive testing measures. Given the prevalence of both sensorineural and conductive hearing loss in NS (Qiu et al. 1998), a referral should be made to an audiologist for further testing if auditory concerns are suspected. A vision assessment may also be appropriate, as eye conditions and refractory errors are well-documented (Lee et al. 1992).
Throughout childhood and adolescence, individuals with NS can have highly variable abilities in areas such as speech/language development, memory, visual processing, and academic skills. As a result, neuropsychological evaluation of individuals with NS must be comprehensive and cover a broad range of domains. Assessment should include measures of processing speed, as this is known to be an area of weakness for many individuals with NS. Ideally, selection of processing speed measures that are less heavily dependent on graphomotor precision (e.g., computerized reaction time, cancellation) could be administered to reduce bias based on fine motor weaknesses. Identification of areas of relative strength in terms of reasoning abilities (e.g., verbal, nonverbal, spatial) will provide avenues for more successful adaptation and learning in the academic environment. It is advisable that assessment of memory examines both visual and verbal memory. Recognition memory should be assessed in addition to recall memory, as a relative strength in recognition memory is seen in many individuals and may provide clues as to optimal teaching and assessment strategies in educational or work settings.
When intellectual deficits and/or hearing loss are present, children with NS are at heightened risk for difficulties in other domains, especially for language and reading disorders. Identification of common comorbidities including ADHD, mathematics or writing disabilities, and social skills concerns (including ASD) should be a focus of assessment. Teacher ratings may be helpful to assess behavior and social skills competence across settings. The impact of peer relationships on the individual’s development and well-being should also be evaluated. It is clear that at least some individuals with NS experience teasing, bullying, or social isolation due to developmental or physical differences, which should be immediately addressed within the environments these experiences are occurring.
In the course of neuropsychological assessment of individuals with NS, collection of collateral information regarding physical health will be essential with regard to both interpretation of test results and formulation of appropriate treatment recommendations. History of preterm birth or birth complications should be assessed, as these factors implicate heightened cognitive and behavioral risk. Seizure history and medication use should be noted. Both hematopoetic malignancies (especially leukemia, which is most often associated with PTPN11 mutations) and malignant solid tumors have been reported in association with NS (Smpokou et al. 2015). For patients with history of cancer, it will be important to assess for late effects of chemotherapy or other cancer treatments. Although cardiac disease does not seem to impose a significant risk factor in terms of intellectual development, the potential psychosocial sequelae of cardiac interventions (e.g., surgeries, hospitalizations, medications) should be considered. In general, children with NS are at heightened risk for medical stress, including feeding and growth issues, endocrine disorders, hematologic concerns (e.g., easy bruising), etc. (Romano et al. 2010). These illness-related experiences should be taken into account when considering social–emotional adjustment.
Special attention should be given to assessment related to the experience of pain. Although the etiology for pain in individuals with NS often not evident, pain does appear to be a relatively common feature among both children and adults. Research has shown that chronic pain in young people has the potential to significantly impact functioning, in terms of increased school absences, reduced involvement in activities, and greater social isolation (Forgeron et al. 2010). Pain experiences could also potentially manifest in terms of heightened distractibility during cognitive or academic tasks. Thus, identification of pain in individuals with NS could affect interpretation of assessment findings. Further, treatment of chronic pain has the potential to greatly impact psychosocial adjustment. In a recent study of adults with NS, chronic muscle, joint, or back pain was reported in the majority of participants (Smpokou et al. 2012). For most individuals, musculoskeletal pain originated in childhood, whereas back pain typically originated in adolescence/adulthood. Several physical aspects of NS may contribute to the occurrence of pain. For example, atypical skeletal features such as chest wall deformity, spinal anomalies, and joint hyper-extensibility are common in NS (Romano et al. 2010). A study of adults reported that over half of participants had been diagnosed with scoliosis in childhood or adolescence (Smpokou et al. 2012). Finally, given a possible predisposition to cerebrovascular disease, hydrocephalus, solid tumors, and other neurological issues in NS (Roberts et al. 2013; Kratz et al. 2011), recurrent severe headaches should prompt a referral to a neurologist.
A final component that may be of great importance in assessment of individuals with NS is the issue of sleep duration and quality. Sleep disorders occur more commonly in children with craniofacial syndromes (Shadfar et al. 2012), and sleep-disordered breathing can lead to adverse effects on the cardiovascular system, neurocognitive function, and growth. Obstructive sleep apnea has been reported in children with NS (Khirani et al. 2012) and has the potential to be life-threatening if untreated. Craniosynostosis has been reported in children with specific mutations in KRAS (Addissie et al. 2015; Kratz et al. 2009) and is a known risk factor for obstructive sleep apnea (Cielo and Marcus 2015). Chiari I malformations can also increase risk for sleep-disordered breathing (Leu 2015; Dauvilliers et al. 2007). Thus, behavioral assessment of individuals with NS should include inquiry regarding potential sleep problems, including difficulty initiating sleep, insufficient sleep duration, restlessness, heavy snoring, increased respiratory effort, or pauses in breathing. A referral for a sleep study should be considered for children with chronic sleep issues.
Educational and Treatment Recommendations
Despite the fact that most individuals with NS do not have significant intellectual impairments, researchers have reported lower levels of educational achievement among adults with NS relative to the general population (Binder et al. 2012). There is some evidence to suggest that individuals who are diagnosed with NS during childhood tend to receive more extensive special education supports compared to individuals who are not diagnosed until adulthood (Noonan 2005). As more individuals are being recognized as having NS at a young age, there are increasing opportunities to intervene and create a more supportive learning environment. It is clear that there are myriad ways in which both physical health aspects of NS and neuropsychological deficits can impact academic functioning and attainment of educational goals. As a result, most individuals with NS would be expected to qualify for special education services to provide support for their learning needs. While special education eligibility can vary across US states, consideration of eligibility for an Individualized Education Plan (IEP) under the category Other Health Disability (or Other Health Impairment) may be appropriate in most situations. Other likely categories under which children with NS may qualify for services include Developmental Delay (for early childhood services), Speech or Language Impairment, Autism Spectrum Disorder, Developmental Cognitive Disability, or Emotional/Behavioral Disorder. Better educational outcomes may be attainable for many individuals with NS with support or services included in an IEP or a Section 504 plan.
In early development, a number of rehabilitation therapies may be needed to address developmental delays and support progress. Due to the hypotonia and motor delays that commonly occur in NS, physical therapy and occupational therapy are frequently appropriate. For children with severe gastrointestinal and feeding issues, referral to a speech/language pathologist or feeding specialist is advised. A variety of issues such as chewing, swallowing, texture sensitivity, and oral aversions may need to be addressed. In situations where feeding difficulties persist past infancy, school accommodations may be necessary to address these concerns. The involvement of nursing staff at school may be important if a child requires tube feeding or has unique nutritional needs. Allowing additional time for consuming food in the lunch room can also be necessary to enable sufficient food intake, as some children with NS experience problems with gagging, difficulty swallowing, slow feeding, or recurrent vomiting (Sharland et al. 1992).
During school-aged years, individualized or small-group special education instruction may be needed to address learning difficulties, depending on the needs of the individual child. Children with NS will often require speech and language therapies to address articulation and language delays. With regard to learning disabilities, some research indicates that children with RASopathies who have reading difficulties may have similar underlying deficits as children with idiopathic reading disabilities (Cutting and Levine 2010). This finding suggests that they may respond to similar types of intervention programs. Therefore, a logical step would be to apply evidence-based treatments for learning disabilities, such as phonics instruction for reading disorder (Galuschka et al. 2014), to instruction for children with NS.
Testing accommodations and modifications of assignment load may be very helpful for some individuals with NS to achieve success. Extended time for classroom work or testing may be appropriate, particularly for children and adults with relative weaknesses in processing speed, attention difficulties, or reading disabilities. Due to difficulties with executive functioning, some children may require help with organization and planning of assignments. Use of multiple modalities in learning and testing will assist children who may have trouble demonstrating retention of information. Preliminary research on learning and memory suggests several possible strategies to enhance learning outcomes. First, some evidence suggests that spreading learning across multiple sessions may enable better memory consolidation (Pagani et al. 2009). Second, providing choices rather than requiring free recall of information could allow children who have difficulty with retrieval or formulation of responses to better demonstrate their knowledge (Pierpont et al. 2013). Finally, presenting information in a meaningful context may be more beneficial as compared with rote learning in individuals with NS.
A number of classroom accommodations may be useful to address physical or developmental issues. The prevalence of short stature among individuals with NS should prompt consideration of ergonomic needs. For example, teachers and occupational therapists should address issues related to desk and chair size and access to equipment and materials. Similarly, some individuals with NS have a webbed neck, which may affect comfort when looking down at a desk and could contribute to pain or headaches. Evaluation by an occupational therapist can assist with optimal positioning (e.g., use of slant boards or consideration of placement of books or writing materials). Fine motor difficulties should also be addressed by appropriate accommodations. If handwriting causes pain or frustration, it may be beneficial to consider a reduced note-taking or handwriting burden. Other fine motor accommodations could include provision of large-lined paper for writing practice, use of enlarged worksheets with extra space, and permitting use of assistive technologies (e.g., use of a keyboard, dictation software). Some children with NS also struggle with balance and coordination issues, so consideration of adapted physical education may be appropriate. Children with congenital heart disease or other medical features may experience poor endurance or fatigue. This difficulty may impact participation in lengthy projects, field trips, or extensive periods of walking. Finally, children with NS may need to be absent from school more than a typical child due to illness or frequent medical appointments. This issue can be addressed in the IEP to ensure that students with NS do not fall behind in their course materials and parents are not targeted related to truancy concerns.
Given that children with NS may experience sensory concerns, these issues may be important to address within the IEP or Section 504 plan. Vision services and adaptations, or work with a hearing specialist, may be necessary for some individuals with visual or hearing impairments. An evaluation related to sensory processing behaviors may also help to identify additional supports. For sensory-related behaviors common in children with comorbid ADHD or ASD diagnoses, consideration of accommodations for noise or light filtering, use of sensory objects (e.g., fidgets, chewable items), or movement breaks may serve the child’s needs.
Importantly, results from surveys of adults with NS indicate that most are able to finish high school and some individuals also attend college (Noonan 2005). For people with NS who attend college or other post-secondary institutions (e.g., vocational or career schools), services such as tutoring programs and academic accommodations should be available through their college’s disability service programs. Indeed, there is reason for great optimism regarding the potential to develop increasingly effective supports and treatments for neuropsychological challenges that may face individuals with NS across the lifespan. It is clear that psychosocial outcomes can be quite positive for many individuals with this diagnosis. Although accommodations and supports will be necessary for many individuals with NS to obtain optimal educational and social success, in many cases, the expectations for personal and professional achievements may not differ greatly as compared with expectations within the general population. While a minority of adults may be unable to work due to disability, most individuals with NS have the capacity to maintain employment (Smpokou et al. 2012), and unemployment rates are generally comparable to the general population (Binder et al. 2012). If an individual does have social or cognitive limitations, these concerns must not always be viewed as a barrier to independent living. Rather, careful planning may assist with very successful transition to adulthood. In this regard, interventions focused on social and practical skills may be helpful for some individuals.
Regarding social–emotional and behavioral adjustment in individuals with NS, several treatment approaches hold promise for improving outcomes. There are currently no published clinical trials to evaluate the effectiveness of pharmacological treatments for mental health issues in NS. However, preliminary research on other RASopathies suggests several avenues for intervention. For example, stimulant medications (e.g., methylphenidate) are known to be effective in improving cognitive, behavioral, and social outcomes in individuals with NF1 who have symptoms of ADHD (Mautner et al. 2002; Lion-Francois et al. 2014). For children with NS, the possibility of medication must be considered in consultation with medical providers familiar with the child’s complex history, lending special attention to growth considerations and cardiac risk. Medication may be also indicated for treatment of anxiety or mood disorders, although additional research is greatly needed in this area to determine the scope and impact of these and other psychiatric conditions in children and adults with NS. Psychotherapeutic and behavioral management interventions should be considered for children with significant emotional or behavioral dysregulation. Cognitive–behavioral therapy is a well-established treatment for childhood depression and anxiety, whereas behavioral parent training requiring involvement of a caregiver may be most effective at addressing disruptive behaviors (Forehand et al. 2013). Children with NS who struggle with social competence may benefit from explicit instruction or counseling to bring them up to par with their peers. Social skills groups have some evidence base for enhancing social outcomes among children with autism spectrum disorders (Reichow et al. 2013), who may share traits with some individuals with NS. Finally, psychological interventions (e.g., cognitive–behavioral therapy, relaxation techniques, mindfulness-based therapy) to target sleep problems or pain/fatigue may be helpful for children with NS who struggle with these issues.
Finally, in recent years, increasing community support is available for individuals with RASopathies. A number of organizations have emerged to provide social support, enable sharing of experiences, support education and research, and provide advocacy (Table 4). These organizations have an integral role to play in fostering knowledge and improving quality of life for individuals with NS and other RASopathies. Parents/caregivers and siblings of individuals with NS can also obtain support through these organizations.
Directions for Future Research
While the literature on neuropsychological features of individuals with NS has greatly increased in recent years, several significant gaps remain. First, in the majority of published studies, a description of neuropsychological outcomes is provided without statistical exploration of possible risk factors that lead to differences in outcome. As genetic testing information becomes increasingly available, the possibility of exploring genotype–phenotype relationships becomes more practicable. Understanding how specific biochemical changes influence brain development will bring us to a better understanding of the role of the RAS pathway in development. In line with this, valuable information will emerge from research that compares individuals with classic NS mutations and phenotype with similar phenotypes (e.g., NSML; NS/LAH; NS phenotype with BRAF or KRAS mutations). Further, understanding the similarities and differences between NS and other distinct RASopathies (e.g., NF1) can also bring us to a better understanding of the variable clinical manifestations of RAS pathway anomalies. Only a handful of articles have evaluated individuals with multiple different RASopathies within the same study (e.g., Alfieri et al. 2014; Adviento et al. 2014; Pierpont et al. 2010b; Cesarini et al. 2009). This type of neuropsychological research can be challenging, as measures that are used to study one population may not easily be applied to other groups due to differences in phenotypic presentation and neurological involvement. Nevertheless, similarities in underlying biochemical substrates and also in clinical needs should motivate more frequent comparisons both among RASopathies and across a variety of medical conditions associated with cognitive and behavioral manifestations (e.g., fragile X syndrome, ASD, ADHD). Moreover, it is essential that studies not only attempt to account for genotypic variability but that they also include other biological, developmental, and environmental factors that may contribute to the broad variation in expression within the same clinical disorder. Variables related to medical severity, perinatal factors, nutrition, and access to services are just a few factors that have the potential to dramatically alter a child’s developmental trajectory and yet have not been extensively studied.
In terms of content area, there are a number of aspects of neuropsychological functioning that have been largely overlooked in NS and the RASopathies. First, there is a lack of longitudinal research to examine how cognitive and behavioral trajectories change over time. This type of research has led to important insights into how neurodevelopmental pathways may differ in children with other genetic conditions such as fragile X syndrome (e.g., Kover et al. 2013; Chromik et al. 2015), leading to insights into how interventions may alter these trajectories. Second, it is clear that more detailed information about the interpersonal experiences of individuals with NS is needed. While some of the research reviewed above suggests that social capacities of individuals with NS frequently differ from the general population, including the ability to form and maintain peer friendships and romantic relationships, this is an area that has received relatively little research attention. Studies examining how differences in social competence, social cognition, or social communication may affect the lives of individuals with NS could provide major benefit for caregivers seeking to provide early intervention opportunities that will facilitate their child’s long-term outcomes. Additionally, environmental contributions to social outcomes, such as exposure to teasing/bullying, should be investigated. Although children with short stature are at heightened risk for teasing, some evidence suggests that teasing due to short stature in isolation may not have clinically meaningful effects on overall psychosocial adjustment (Sandberg 2011). Additional research into social experiences of individuals with NS would help elucidate whether there is a significant impact of teasing/victimization in this group.
Translational research to inform what types of treatments could be safe and effective is in its early stages. In particular, great interest has developed with regard to interventions that might improve cognition, learning, and memory in the RASopathies (Shilyansky et al. 2010b). Studies involving animal models of NF1 (e.g., Costa et al. 2002) have led to the first clinical trials that target abnormal Ras signaling using statin drugs. Phase 1 trials indicate that these drugs can be safely prescribed in children with NF1, and there is some evidence that the drug lovastatin could affect specific cognitive outcomes (Acosta et al. 2011). However, results of initial trials using simvastatin did not support short-term efficacy in providing clear functional benefits (Krab et al. 2008; van der Vaart et al. 2013). NF1 treatment trials to evaluate statin drugs using larger, more carefully selected samples are ongoing (Acosta et al. 2012). Importantly, there is now evidence to demonstrate that drugs targeting the same mechanism can be effective in ameliorating cognitive deficits in mouse models of NS (Lee et al. 2014), suggesting a potential avenue for future therapeutic exploration.
Some researchers have advised that to optimally translate scientific advances into clinical interventions, a collaborative framework to include neuroscientists and clinicians from diverse institutions must be developed (Acosta et al. 2012). The complexity of the challenges facing evaluation of clinical therapies (e.g., measurement considerations, recruitment of sufficient numbers of participants) will require interdisciplinary contributions and multi-site partnerships. For some populations, these types of collaborations to identify appropriate study designs and endpoints for clinical trials are underway (e.g., the Response Evaluation in Neurofibromatosis and Schwannomatosis (REiNS) collaborative groups; Plotkin et al. 2013). Finally, alongside pharmacological treatments, it will also be critical to examine the efficacy of behavioral and family interventions. While drug treatments hold promise for improving cognitive or self-regulatory mechanisms, they will not address skill deficits or other psychosocial adjustment issues related to medical or interpersonal experiences. With the support of patient advocacy organizations and funding agencies, development of these types of treatments is also attainable. Indeed, NS and the RASopathies present an extremely unique opportunity to study the effects of specific biochemical processes on brain and behavior and to identify ways to improve clinical care and mental health outcomes for individuals with genetic differences.
REFERENCES
Acosta, M. T., Kardel, P. G., Walsh, K. S., Rosenbaum, K. N., Gioia, G. A., & Packer, R. J. (2011). Lovastatin as treatment for neurocognitive deficits in neurofibromatosis type 1: phase I study. Pediatric Neurology, 45(4), 241–245. doi:10.1016/j.pediatrneurol.2011.06.016.
Acosta, M. T., Bearden, C. E., Castellanos, F. X., Cutting, L., Elgersma, Y., Gioia, G., et al. (2012). The Learning Disabilities Network (LeaDNet): using neurofibromatosis type 1 (NF1) as a paradigm for translational research. American Journal of Medical Genetics. Part A, 158A(9), 2225–2232. doi:10.1002/ajmg.a.35535.
Addissie, Y. A., Kotecha, U., Hart, R. A., Martinez, A. F., Kruszka, P., & Muenke, M. (2015). Craniosynostosis and Noonan syndrome with KRAS mutations: expanding the phenotype with a case report and review of the literature. American Journal of Medical Genetics. Part A. doi:10.1002/ajmg.a.37259.
Adviento, B., Corbin, I. L., Widjaja, F., Desachy, G., Enrique, N., Rosser, T., et al. (2014). Autism traits in the RASopathies. Journal of Medical Genetics, 51(1), 10–20. doi:10.1136/jmedgenet-2013-101951.
Alfieri, P., Cesarini, L., Zampino, G., Pantaleoni, F., Selicorni, A., Salerni, A., et al. (2008). Visual function in Noonan and LEOPARD syndrome. Neuropediatrics, 39(6), 335–340. doi:10.1055/s-0029-1216354.
Alfieri, P., Cesarini, L., De Rose, P., Ricci, D., Selicorni, A., Menghini, D., et al. (2011a). Visual processing in Noonan syndrome: dorsal and ventral stream sensitivity. American Journal of Medical Genetics. Part A, 155A(10), 2459–2464. doi:10.1002/ajmg.a.34229.
Alfieri, P., Cesarini, L., Mallardi, M., Piccini, G., Caciolo, C., Leoni, C., et al. (2011b). Long term memory profile of disorders associated with dysregulation of the RAS-MAPK signaling cascade. Behavior Genetics, 41(3), 423–429. doi:10.1007/s10519-011-9446-5.
Alfieri, P., Piccini, G., Caciolo, C., Perrino, F., Gambardella, M. L., Mallardi, M., et al. (2014). Behavioral profile in RASopathies. American Journal of Medical Genetics. Part A. doi:10.1002/ajmg.a.36374.
Allanson, J. E., Anneren, G., Aoki, Y., Armour, C. M., Bondeson, M. L., Cave, H., et al. (2011). Cardio-facio-cutaneous syndrome: does genotype predict phenotype? American Journal of Medical Genetics. Part C, 157(2), 129–135. doi:10.1002/ajmg.c.30295.
American Psychiatric Association (2000). Diagnostic and statistical manual of mental disorders, fourth edition-text revision. Washington, DC: Author.
Aoki, Y., Niihori, T., Banjo, T., Okamoto, N., Mizuno, S., Kurosawa, K., et al. (2013). Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. American Journal of Human Genetics, 93(1), 173–180. doi:10.1016/j.ajhg.2013.05.021.
Arvaniti, A., Samakouri, M., Keskeridou, F., & Veletza, S. (2014). Concurrence of anorexia nervosa and Noonan syndrome. European Eating Disorders Review, 22(1), 83–85. doi:10.1002/erv.2261.
Bertola, D. R., Pereira, A. C., de Oliveira, P. S., Kim, C. A., & Krieger, J. E. (2004). Clinical variability in a Noonan syndrome family with a new PTPN11 gene mutation. American Journal of Medical Genetics. Part A, 130A(4), 378–383. doi:10.1002/ajmg.a.30270.
Binder, G., Grathwol, S., von Loeper, K., Blumenstock, G., Kaulitz, R., Freiberg, C., et al. (2012). Health and quality of life in adults with Noonan syndrome. The Journal of Pediatrics, 161(3), 501–505. doi:10.1016/j.jpeds.2012.02.043. e501.
Cesarini, L., Alfieri, P., Pantaleoni, F., Vasta, I., Cerutti, M., Petrangeli, V., et al. (2009). Cognitive profile of disorders associated with dysregulation of the RAS/MAPK signaling cascade. American Journal of Medical Genetics. Part A, 149A(2), 140–146. doi:10.1002/ajmg.a.32488.
Chromik, L. C., Quintin, E. M., Lepage, J. F., Hustyi, K. M., Lightbody, A. A., & Reiss, A. L. (2015). The influence of hyperactivity, impulsivity, and attention problems on social functioning in adolescents and young adults with fragile X syndrome. Journal of Attention Disorders. doi:10.1177/1087054715571739.
Cielo, C. M., & Marcus, C. L. (2015). Obstructive sleep apnoea in children with craniofacial syndromes. Paediatric Respiratory Reviews, 16(3), 189–196. doi:10.1016/j.prrv.2014.11.003.
Colquitt, J. L., & Noonan, J. A. (2014). Cardiac findings in Noonan syndrome on long-term follow-up. Congenital Heart Disease, 9(2), 144–150. doi:10.1111/chd.12102.
Constantino, J., & Gruber, C. (2005). The Social Responsiveness Scale Manual. Los Angeles, CA: Western Psychological Services.
Cordeddu, V., Di Schiavi, E., Pennacchio, L. A., Ma'ayan, A., Sarkozy, A., Fodale, V., et al. (2009). Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nature Genetics, 41(9), 1022–1026. doi:10.1038/ng.425.
Costa, R. M., Yang, T., Huynh, D. P., Pulst, S. M., Viskochil, D. H., Silva, A. J., et al. (2001). Learning deficits, but normal development and tumor predisposition, in mice lacking exon 23a of Nf1. Nature Genetics, 27(4), 399–405. doi:10.1038/86898.
Costa, R. M., Federov, N. B., Kogan, J. H., Murphy, G. G., Stern, J., Ohno, M., et al. (2002). Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature, 415(6871), 526–530. doi:10.1038/nature711.
Cutting, L. E., & Levine, T. M. (2010). Cognitive profile of children with neurofibromatosis and reading disabilities. Child Neuropsychology, 16(5), 417–432. doi:10.1080/09297041003761985.
Dauvilliers, Y., Stal, V., Abril, B., Coubes, P., Bobin, S., Touchon, J., et al. (2007). Chiari malformation and sleep related breathing disorders. Journal of Neurology, Neurosurgery and Psychiatry, 78(12), 1344–1348. doi:10.1136/jnnp.2006.108779.
Davis, S., & Laroche, S. (2006). Mitogen-activated protein kinase/extracellular regulated kinase signalling and memory stabilization: a review. Genes, Brain, and Behavior, 5(2), 61–72. doi:10.1111/j.1601-183X.2006.00230.x.
Diggs-Andrews, K. A., & Gutmann, D. H. (2013). Modeling cognitive dysfunction in neurofibromatosis-1. Trends in Neurosciences, 36(4), 237–247. doi:10.1016/j.tins.2012.12.002.
Elliott, C. (1990). Differential Ability Scales. San Antonio, TX: The Psychological Corporation.
Forehand, R., Jones, D. J., & Parent, J. (2013). Behavioral parenting interventions for child disruptive behaviors and anxiety: what's different and what's the same. Clinical Psychology Review, 33(1), 133–145. doi:10.1016/j.cpr.2012.10.010.
Forgeron, P. A., King, S., Stinson, J. N., McGrath, P. J., MacDonald, A. J., & Chambers, C. T. (2010). Social functioning and peer relationships in children and adolescents with chronic pain: a systematic review. Pain Research and Management, 15(1), 27–41.
Galasso, C., Lo-Castro, A., Di Carlo, L., Pitzianti, M. B., D'Agati, E., Curatolo, P., et al. (2014). Planning deficit in children with neurofibromatosis type 1: a neurocognitive trait independent from attention-deficit hyperactivity disorder (ADHD)? Journal of Child Neurology. doi:10.1177/0883073813517001.
Galuschka, K., Ise, E., Krick, K., & Schulte-Korne, G. (2014). Effectiveness of treatment approaches for children and adolescents with reading disabilities: a meta-analysis of randomized controlled trials. PloS One, 9(2), e89900. doi:10.1371/journal.pone.0089900.
Gauthier, A. S., Furstoss, O., Araki, T., Chan, R., Neel, B. G., Kaplan, D. R., et al. (2007). Control of CNS cell-fate decisions by SHP-2 and its dysregulation in Noonan syndrome. Neuron, 54(2), 245–262. doi:10.1016/j.neuron.2007.03.027.
Griffiths, R., & Huntley, M. (1996). The Griffiths Mental Development Scales: from birth to 2 years. The Test Agency Ltd: Henley-on-Thames.
Gripp, K. W., Zand, D. J., Demmer, L., Anderson, C. E., Dobyns, W. B., Zackai, E. H., et al. (2013). Expanding the SHOC2 mutation associated phenotype of Noonan syndrome with loose anagen hair: structural brain anomalies and myelofibrosis. American Journal of Medical Genetics. Part A, 161A(10), 2420–2430. doi:10.1002/ajmg.a.36098.
Isenberg, J. C., Templer, A., Gao, F., Titus, J. B., & Gutmann, D. H. (2012). Attention skills in children with neurofibromatosis type 1. Journal of Child Neurology. doi:10.1177/0883073812439435.
Keh, Y. S., Abernethy, L., & Pettorini, B. (2013). Association between Noonan syndrome and Chiari I malformation: a case-based update. Child's Nervous System, 29(5), 749–752. doi:10.1007/s00381-012-2000-9.
Khirani, S., Leboulanger, N., Ramirez, A., & Fauroux, B. (2012). Life-threatening obstructive sleep apnea caused by adenoid hypertrophy in an infant with Noonan syndrome. Case Reports in Pediatrics, 2012, 514514. doi:10.1155/2012/514514.
Koudova, M., Seemanova, E., & Zenker, M. (2009). Novel BRAF mutation in a patient with LEOPARD syndrome and normal intelligence. European Journal of Medical Genetics, 52(5), 337–340. doi:10.1016/j.ejmg.2009.04.006.
Kover, S. T., Pierpont, E. I., Kim, J. S., Brown, W. T., & Abbeduto, L. (2013). A neurodevelopmental perspective on the acquisition of nonverbal cognitive skills in adolescents with fragile X syndrome. Developmental Neuropsychology, 38(7), 445–460. doi:10.1080/87565641.2013.820305.
Krab, L. C., de Goede-Bolder, A., Aarsen, F. K., Pluijm, S. M., Bouman, M. J., van der Geest, J. N., et al. (2008). Effect of simvastatin on cognitive functioning in children with neurofibromatosis type 1: a randomized controlled trial. Journal of the American Medical Association, 300(3), 287–294. doi:10.1001/jama.300.3.287.
Kratz, C. P., Zampino, G., Kriek, M., Kant, S. G., Leoni, C., Pantaleoni, F., et al. (2009). Craniosynostosis in patients with Noonan syndrome caused by germline KRAS mutations. American Journal of Medical Genetics. Part A, 149A(5), 1036–1040. doi:10.1002/ajmg.a.32786.
Kratz, C. P., Rapisuwon, S., Reed, H., Hasle, H., & Rosenberg, P. S. (2011). Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. American Journal of Medical Genetics. Part C, 157C(2), 83–89. doi:10.1002/ajmg.c.30300.
Lee, N. B., Kelly, L., & Sharland, M. (1992). Ocular manifestations of Noonan syndrome. Eye, 6(Pt 3), 328–334. doi:10.1038/eye.1992.66.
Lee, D. A., Portnoy, S., Hill, P., Gillberg, C., Patton, M. A., Allanson, J., et al. (2005). Psychological profile of children with Noonan syndrome. Developmental Medicine and Child Neurology, 47(1), 35–38.
Lee, B. H., Kim, J. M., Jin, H. Y., Kim, G. H., Choi, J. H., & Yoo, H. W. (2011). Spectrum of mutations in Noonan syndrome and their correlation with phenotypes. The Journal of Pediatrics, 159(6), 1029–1035. doi:10.1016/j.jpeds.2011.05.024.
Lee, Y. S., Ehninger, D., Zhou, M., Oh, J. Y., Kang, M., Kwak, C., et al. (2014). Mechanism and treatment for learning and memory deficits in mouse models of Noonan syndrome. Nature Neuroscience, 17(12), 1736–1743. doi:10.1038/nn.3863.
Leu, R. M. (2015). Sleep related breathing disorders and the Chiari 1 malformation. Chest. doi:10.1378/chest.14-3090.
Lindstrom, K., Winbladh, B., Haglund, B., & Hjern, A. (2007). Preterm infants as young adults: a Swedish national cohort study. Pediatrics, 120(1), 70–77. doi:10.1542/peds.2006-3260.
Lion-Francois, L., Gueyffier, F., Mercier, C., Gerard, D., Herbillon, V., Kemlin, I., et al. (2014). The effect of methylphenidate on neurofibromatosis type 1: a randomised, double-blind, placebo-controlled, crossover trial. Orphanet Journal of Rare Diseases, 9(1), 142. doi:10.1186/s13023-014-0142-4.
Lo-Castro, A., D'Agati, E., & Curatolo, P. (2011). ADHD and genetic syndromes. Brain and Development, 33(6), 456–461. doi:10.1016/j.braindev.2010.05.011.
Luckasson, R., Borthwick-Duffy, S., Buntinx, W. H. E., Coulter, D. L., Craig, E. M., Schalock, R. L., et al. (2002). Mental retardation: definition, classification, and systems of supports (10th ed.). Washington, DC US: American Association on Mental Retardation.
Mainberger, F., Zenker, M., Jung, N. H., Delvendahl, I., Brandt, A., Freudenberg, L., et al. (2013). Impaired motor cortex plasticity in patients with Noonan syndrome. Clinical Neurophysiology, 124(12), 2439–2444. doi:10.1016/j.clinph.2013.04.343.
Marin, L., da Silva, F. T., de Sa, L. C., Brasil, A. S., Pereira, A., Furquim, I. M., et al. (2012). Ocular manifestations of Noonan syndrome. Ophthalmic Genetics, 33(1), 1–5. doi:10.3109/13816810.2011.593606.
Marino, B. S., Lipkin, P. H., Newburger, J. W., Peacock, G., Gerdes, M., Gaynor, J. W., et al. (2012). Neurodevelopmental outcomes in children with congenital heart disease: evaluation and management: a scientific statement from the American Heart Association. Circulation, 126(9), 1143–1172. doi:10.1161/CIR.0b013e318265ee8a.
Mautner, V. F., Kluwe, L., Thakker, S. D., & Leark, R. A. (2002). Treatment of ADHD in neurofibromatosis type 1. Developmental Medicine and Child Neurology, 44(3), 164–170.
Mendez, H. M., & Opitz, J. M. (1985). Noonan syndrome: a review. American Journal of Medical Genetics, 21(3), 493–506. doi:10.1002/ajmg.1320210312.
Moster, D., Lie, R. T., & Markestad, T. (2008). Long-term medical and social consequences of preterm birth. The New England Journal of Medicine, 359(3), 262–273. doi:10.1056/NEJMoa0706475.
Neri, G., Allanson, J., & Kavamura, M. I. (2008). No reason yet to change diagnostic criteria for Noonan, Costello and cardio-facio-cutaneous syndromes. Journal of Medical Genetics, 45(12), 832. doi:10.1136/jmg.2008.063263.
Nishi, E., Mizuno, S., Nanjo, Y., Niihori, T., Fukushima, Y., Matsubara, Y., et al. (2015). A novel heterozygous MAP2K1 mutation in a patient with Noonan syndrome with multiple lentigines. American Journal of Medical Genetics. Part A, 167(2), 407–411. doi:10.1002/ajmg.a.36842.
Noonan, J. A. (2005). Noonan syndrome. In S. Goldstein & C. R. Reynolds (Eds.), Handbook of neurodevelopmental and genetic disorders in adults (pp. 308–319). New York: Guilford Press.
Noonan Syndrome Guideline Development Group (2010). Management of Noonan syndrome: a clinical guideline. Resource document. Rasopathies Network. https://rasopathiesnet.org/wp-content/uploads/2014/01/265_Noonan_Guidelines.pdf. Accessed 8 Aug 2015.
Noonan, J. A., & Ehmke, D. A. (1963). Associated noncardiac malformations in children with congenital heart disease. Journal of Pediatrics, 63, 468–470.
Nystrom, A. M., Ekvall, S., Berglund, E., Bjorkqvist, M., Braathen, G., Duchen, K., et al. (2008). Noonan and cardio-facio-cutaneous syndromes: two clinically and genetically overlapping disorders. Journal of Medical Genetics, 45, 500–506. doi:10.1136/jmg.2008.057653.
Pagani, M. R., Oishi, K., Gelb, B. D., & Zhong, Y. (2009). The phosphatase SHP2 regulates the spacing effect for long-term memory induction. Cell, 139(1), 186–198. doi:10.1016/j.cell.2009.08.033.
Payne, J. M., Hyman, S. L., Shores, E. A., & North, K. N. (2011). Assessment of executive function and attention in children with neurofibromatosis type 1: relationships between cognitive measures and real-world behavior. Child Neuropsychology, 17(4), 313–329. doi:10.1080/09297049.2010.542746.
Pierpont, E. I., Pierpont, M. E., Mendelsohn, N. J., Roberts, A. E., Tworog-Dube, E., & Seidenberg, M. S. (2009). Genotype differences in cognitive functioning in Noonan syndrome. Genes, Brain and Behavior, 8(3), 275–282. doi:10.1111/j.1601-183X.2008.00469.x.
Pierpont, E. I., Ellis Weismer, S., Roberts, A. E., Tworog-Dube, E., Pierpont, M. E., Mendelsohn, N. J., et al. (2010a). The language phenotype of children and adolescents with Noonan syndrome. Journal of Speech, Language, and Hearing Research, 53(4), 917–932. doi:10.1044/1092-4388(2009/09-0046).
Pierpont, E. I., Pierpont, M. E., Mendelsohn, N. J., Roberts, A. E., Tworog-Dube, E., Rauen, K. A., et al. (2010b). Effects of germline mutations in the Ras/MAPK signaling pathway on adaptive behavior: cardiofaciocutaneous syndrome and Noonan syndrome. American Journal of Medical Genetics. Part A, 152A(3), 591–600. doi:10.1002/ajmg.a.33268.
Pierpont, E. I., Tworog-Dube, E., & Roberts, A. E. (2013). Learning and memory in children with Noonan syndrome. American Journal of Medical Genetics. Part A, 161(9), 2250–2257. doi:10.1002/ajmg.a.36075.
Pierpont, E. I., Tworog-Dube, E., & Roberts, A. E. (2015). Attention skills and executive functioning in children with Noonan syndrome and their unaffected siblings. Developmental Medicine and Child Neurology, 57(4), 385–392. doi:10.1111/dmcn.12621.
Piers, E., & Harris, D. (1989). The Pier-Harris Children's Self-Concept Scale. Los Angeles, CA: Western Psychological Services.
Plotkin, S. R., Blakeley, J. O., Dombi, E., Fisher, M. J., Hanemann, C. O., Walsh, K. S., et al. (2013). Achieving consensus for clinical trials: the REiNS International Collaboration. Neurology, 81(21 Suppl 1), S1–5. doi:10.1212/01.wnl.0000435743.49414.b6.
Powell, K. B., & Voeller, K. K. (2004). Prefrontal executive function syndromes in children. Journal of Child Neurology, 19(10), 785–797.
Qiu, W., Yin, S., & Stucker, F. (1998). Audiologic manifestations of Noonan syndrome. Otolaryngology - Head and Neck Surgery, 118(3, Pt. 1), 319–323.
Rauen, K. A. (2013). The RASopathies. Annual Review of Genomics and Human Genetics, 14, 355–369. doi:10.1146/annurev-genom-091212-153523.
Reichow, B., Steiner, A. M., & Volkmar, F. (2013). Cochrane review: social skills groups for people aged 6 to 21 with autism spectrum disorders (ASD). Evidence-based Child Health, 8(2), 266–315. doi:10.1002/ebch.1903.
Rey, A. (1941). L'examen psychologique dans les cas d'encephalopathie traumatique. Archives de Psychologie, 28, 286–340.
Roberts, A. E., Araki, T., Swanson, K. D., Montgomery, K. T., Schiripo, T. A., Joshi, V. A., et al. (2007). Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nature Genetics, 39(1), 70–74. doi:10.1038/ng1926.
Roberts, A. E., Allanson, J. E., Tartaglia, M., & Gelb, B. D. (2013). Noonan syndrome. Lancet, 381(9863), 333–342. doi:10.1016/S0140-6736(12)61023-X.
Robins, D., Fein, D., Barton, M., & Green, J. (2001). The Modified Checklist for Autism in Toddlers: an initial study investigating the early detection of autism and pervasive developmental disorders. Journal of Autism and Developmental Disorders, 31, 131–144.
Romano, A. A., Allanson, J. E., Dahlgren, J., Gelb, B. D., Hall, B., Pierpont, M. E., et al. (2010). Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics, 126(4), 746–759. doi:10.1542/peds.2009-3207.
Rowbotham, I., Pit-ten Cate, I. M., Sonuga-Barke, E. J., & Huijbregts, S. C. (2009). Cognitive control in adolescents with neurofibromatosis type 1. Neuropsychology, 23(1), 50–60. doi:10.1037/a0013927.
Rutter, M., Bailey, A., & Lord, C. (2003). Social communication questionnaire. Los Angeles, CA: Western Psychological Services.
Samuels, I. S., Saitta, S. C., & Landreth, G. E. (2009). MAP'ing CNS development and cognition: an ERKsome process. Neuron, 61(2), 160–167. doi:10.1016/j.neuron.2009.01.001.
Sandberg, D. E. (2011). Short stature: psychosocial interventions. Hormone Research in Pædiatrics, 76(Suppl 3), 29–32. doi:10.1159/000330151.
Sarimski, K. (2000). Developmental and behavioural phenotype in Noonan syndrome? Genetic Counseling, 11(4), 383–390.
Sarkozy, A., Digilio, M. C., & Dallapiccola, B. (2008). LEOPARD syndrome. Orphanet Journal of Rare Diseases, 3, 13. doi:10.1186/1750-1172-3-13.
Sarkozy, A., Carta, C., Moretti, S., Zampino, G., Digilio, M. C., Pantaleoni, F., et al. (2009). Germline BRAF mutations in Noonan, LEOPARD, and cardiofaciocutaneous syndromes: molecular diversity and associated phenotypic spectrum. Human Mutation, 30(4), 695–702. doi:10.1002/humu.20955.
Scantlebury, N., Cunningham, T., Dockstader, C., Laughlin, S., Gaetz, W., Rockel, C., et al. (2014). Relations between white matter maturation and reaction time in childhood. Journal of the International Neuropsychological Society, 20(1), 99–112. doi:10.1017/S1355617713001148.
Schubbert, S., Zenker, M., Rowe, S., Boll, S., Klein, C., Bollag, G., et al. (2006). Germline KRAS mutations cause Noonan syndrome. Nature Genetics, 38(3), 331–336. doi:10.1038/ng1748.
Shadfar, S., Drake, A. F., Vaughn, B. V., & Zdanski, C. J. (2012). Pediatric airway abnormalities: evaluation and management. Oral and Maxillofacial Surgery Clinics of North America, 24(3), 325–336. doi:10.1016/j.coms.2012.04.005.
Shah, N., Rodriguez, M., Louis, D. S., Lindley, K., & Milla, P. J. (1999). Feeding difficulties and foregut dysmotility in Noonan's syndrome. Archives of Disease in Childhood, 81(1), 28–31.
Sharland, M., Burch, M., McKenna, W., & Paton, M. (1992). A clinical study of Noonan Syndrome. Archives of Disease in Childhood, 67(2), 178–183.
Shaw, A. C., Kalidas, K., Crosby, A. H., Jeffery, S., & Patton, M. A. (2007). The natural history of Noonan syndrome: a long-term follow-up study. Archives of Disease in Childhood, 92(2), 128–132. doi:10.1136/adc.2006.104547.
Sheslow, D., & Adams, W. (2003). Wide range assessment of memory and learning (2nd ed.). Lutz, FL: Psychological Assessment Resources, Inc.
Shilyansky, C., Karlsgodt, K. H., Cummings, D. M., Sidiropoulou, K., Hardt, M., James, A. S., et al. (2010a). Neurofibromin regulates corticostriatal inhibitory networks during working memory performance. Proceedings of the National Academy of Sciences of the United States of America, 107(29), 13141–13146. doi:10.1073/pnas.1004829107.
Shilyansky, C., Lee, Y. S., & Silva, A. J. (2010b). Molecular and cellular mechanisms of learning disabilities: a focus on NF1. Annual Review of Neuroscience, 33, 221–243. doi:10.1146/annurev-neuro-060909-153215.
Smpokou, P., Tworog-Dube, E., Kucherlapati, R. S., & Roberts, A. E. (2012). Medical complications, clinical findings, and educational outcomes in adults with Noonan syndrome. American Journal of Medical Genetics. Part A, 158A(12), 3106–3111. doi:10.1002/ajmg.a.35639.
Smpokou, P., Zand, D. J., Rosenbaum, K. N., & Summar, M. L. (2015). Malignancy in Noonan syndrome and related disorders. Clinical Genetics. doi:10.1111/cge.12568.
Storch, E. A., & Ledley, D. R. (2005). Peer victimization and psychosocial adjustment in children: current knowledge and future directions. Clinical Pediatrics, 44(1), 29–38.
Tartaglia, M., Mehler, E. L., Goldberg, R., Zampino, G., Brunner, H. G., Kremer, H., et al. (2001). Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nature Genetics, 29(4), 465–468. doi:10.1038/ng772.
Tartaglia, M., Kalidas, K., Shaw, A., Song, X., Musat, D. L., van der Burgt, I., et al. (2002). PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. American Journal of Human Genetics, 70(6), 1555–1563. doi:10.1086/340847.
Tartaglia, M., Pennacchio, L. A., Zhao, C., Yadav, K. K., Fodale, V., Sarkozy, A., et al. (2007). Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nature Genetics, 39(1), 75–79. doi:10.1038/ng1939.
Tartaglia, M., Gelb, B. D., & Zenker, M. (2011). Noonan syndrome and clinically related disorders. Best practice and research. Clinical Endocrinology and Metabolism, 25(1), 161–179. doi:10.1016/j.beem.2010.09.002.
Tiffen, J. (1968). Purdue pegboard test. Chicago: Science Research Associates.
van der Burgt, I., Thoonen, G., Roosenboom, N., Assman-Hulsmans, C., Gabreels, F., Otten, B., et al. (1999). Patterns of cognitive functioning in school-aged children with Noonan syndrome associated with variability in phenotypic expression. Journal of Pediatrics, 135(6), 707–713.
van der Steene, G., van Haasen, P., de Bruyn, E., Coetsier, P., Pijl, Y., Poortinga, Y., et al. (1986). Wechsler Intelligence Scale for Children-Revised (Dutch version). Lisse: Swets & Zeitlinger.
van der Vaart, T., Plasschaert, E., Rietman, A. B., Renard, M., Oostenbrink, R., Vogels, A., et al. (2013). Simvastatin for cognitive deficits and behavioural problems in patients with neurofibromatosis type 1 (NF1-SIMCODA): a randomised, placebo-controlled trial. Lancet Neurology, 12(11), 1076–1083. doi:10.1016/S1474-4422(13)70227-8.
Verhoeven, W., Wingbermuhle, E., Egger, J., Van der Burgt, I., & Tuinier, S. (2008). Noonan syndrome: psychological and psychiatric aspects. American Journal of Medical Genetics. Part A, 146A(2), 191–196. doi:10.1002/ajmg.a.32115
Wechsler, D. (1974). Wechsler Intelligence Scale for Children, Revised. New York: Psychological Corporation.
Wechsler, D. (1989). Wechsler Preschool and Primary Scale of Intelligence, Revised. San Antonio, TX: The Psychological Corporation.
Wechsler, D. (1997). Wechsler Adult Intelligence Scale (3rd ed.). San Antonio, TX: Psychological Corporation.
Wechsler, D. (1999). Wechsler Abbreviated Scale of Intelligence. San Antonio: Pearson.
Willcutt, E., Sonuga-Barke, E., Nigg, J., & Sergeant, J. (2008). Recent developments in neuropsychological models of childhood psychiatric disorders. In T. Banaschewski, & L. Rohde (Eds.), Biological child psychiatry. Recent trends and developments (Vol. 24, pp. 195–226, Advances in Biological Psychiatry). Basel: Karger.
Wingbermühle, E., Egger, J., van der Burgt, I., & Verhoeven, W. (2009). Neuropsychological and behavioral aspects of Noonan syndrome. Hormone Research, 72(Suppl 2), 15–23. doi:10.1159/000243774.
Wingbermühle, E., Egger, J. I., Verhoeven, W. M., van der Burgt, I., & Kessels, R. P. (2012a). Affective functioning and social cognition in Noonan syndrome. Psychological Medicine, 42, 419–426. doi:10.1017/S0033291711001115.
Wingbermühle, E., Roelofs, R. L., van der Burgt, I., Souren, P. M., Verhoeven, W. M., Kessels, R. P., et al. (2012b). Cognitive functioning of adults with Noonan syndrome: a case-control study. Genes, Brain and Behavior, 11(7), 785–793. doi:10.1111/j.1601-183X.2012.00821.x.
Wood, A., Massarano, A., Super, M., & Harrington, R. (1995). Behavioural aspects and psychiatric findings in Noonan's syndrome. Archives of Disease in Childhood, 72(2), 153–155.
Yoon, G., Rosenberg, J., Blaser, S., & Rauen, K. A. (2007). Neurological complications of cardio-facio-cutaneous syndrome. Developmental Medicine and Child Neurology, 49(12), 894–899. doi:10.1111/j.1469-8749.2007.00894.x.
Zarate, Y. A., Lichty, A. W., Champion, K. J., Clarkson, L. K., Holden, K. R., & Matheus, M. G. (2013). Unique cerebrovascular anomalies in Noonan syndrome with RAF1 mutation. Journal of Child Neurology, 29(8), NP13–NP17. doi:10.1177/0883073813492384.
Acknowledgments
The author wishes to express gratitude to the families with NS who have devoted their time and efforts to participation in research studies. Special thanks to Dr. Margaret Semrud-Clikeman, Kelly Mayr, and Ann Yurcek for their helpful review and suggestions. I am also grateful for the support provided by Dr. Richard Ziegler and the Kristine Nelson Charitable Foundation in facilitating the completion of this manuscript.
Compliance with Ethical Standards
The author declares no conflicts of interest with regard to this manuscript. No research with human participants or animals was carried out in preparation of this review paper.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Pierpont, E.I. Neuropsychological Functioning in Individuals with Noonan Syndrome: a Systematic Literature Review with Educational and Treatment Recommendations. J Pediatr Neuropsychol 2, 14–33 (2016). https://doi.org/10.1007/s40817-015-0005-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40817-015-0005-5