
Abstract
Coronavirus disease 2019 (COVID-19) accounts for over 180,000 deaths in the USA. Although COVID-19 affects all racial ethnicities, non-Hispanic Blacks have the highest mortality rates. Evidence continues to emerge, linking the disproportion of contagion and mortality from severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), a result of adverse social determinants of health. Yet, genetic predisposition may also play a credible role in disease transmission. SARS-CoV-2 enters cells by interaction between SARS-CoV-2 spike protein and the receptor molecule angiotensin converting enzyme 2 (ACE2) expressed on the surface of the target cells, such that polymorphisms and the expression level of ACE2 influence infectivity and consequent pathogenesis of SARS-CoV-2. Genetic polymorphisms in other multiple genes, such as acetylcholinesterase (AChE) and interleukin-6, are also closely associated with underlying diseases, such as hypertension and type 2 diabetes mellitus, which substantially raise SARS-CoV-2 mortality. However, it is unknown how these genetic polymorphisms contribute to the disparate mortality rates, with or without underlying diseases. Of particular interest is the potential that genetic polymorphisms in these genes may be influencing the disparity of COVID-19 mortality rates in Black communities. Here, we review the evidence that biological predisposition for high-risk comorbid conditions may be relevant to our ability to fully understand and therefore address health disparities of COVID-19 deaths in Blacks.
Similar content being viewed by others
Introduction
Health disparities in the COVID-19 pandemic are glaringly apparent, which is due in part to the increased prevalence of comorbidities affecting minority racial/ethnic groups including Blacks, Hispanics, Native Americans, and Asian Americans [1,2,3,4,5]. To date, greater than 180,000 deaths have occurred in the USA, having the greatest mortality among Non-Hispanic Blacks (NHBs) compared with any other racial group [6]. In Chicago, NHBs comprise only 30% of the population. Yet, NHBs represent 72% of the deaths related to COVID-19. Similarly, in Michigan, Milwaukee, Minnesota, and Louisiana, Blacks are dying at higher rates than Whites [6]. Such statistics are not surprising given the inequities of health outcomes confronting NHBs and other minority populations. As adverse social determinants of health (SDH; e.g., socioeconomic status, access to healthcare, physical environment, social support networks, education) perpetuate the disproportion of deaths caused by COVID-19, so will the known risk for their complications due to pre-existing comorbid disease become evident.
SDH and chronic stress certainly alter biological responses in a manner that increases risk for chronic disease [7, 8]. Increased cortisol levels produced by chronic psychological stress have been associated with the onset and progression of chronic inflammatory disease [9]. Underlying conditions independent of stress also mediate pathophysiologic mechanisms of inflammatory disease. For example, in individuals with metabolic syndrome, excess body fat called “overfat” drives chronic IL-6-mediated inflammation in type II diabetes mellitus and (T2DM) and hypertension (HTN) [10,11,12]. These and other pathways of immune-mediated inflammatory disease are in contrast to infection-related cytokine storms (refs). It is also well understood that genetic variants with different allele frequencies in human populations are a result of a group’s ancestral history, some of which have direct relevance to human health and population-specific risk for disease [13]. Together, assuming that such factors are revealed, what remains as a critical gap is a thorough understanding of the complex interactions between the sociological, psychological, and biological human domains that alter disease susceptibility. At the individual level, the intersections between biological and behavioral domains of influence are considered important to expand our knowledge of the complexity in defining health determinants across race. Genomic risk, epigenetic factors, and race are revealing opportunities for new breakthroughs to collectively address sources of risk and develop targeted interventions alongside existing social determinants of health. Specifically, Fig. 1 illustrates a conceptual model through which psychological influences (stress, anxiety, depression), pre-existing/comorbid disease (e.g., HTN, T2DM), and COVID-19 interconnect on the basis of known and unknown genetic variations that translate into human health outcomes and molecular modes of viral pathogenesis. Here, we discuss the evidence that genetic and biological risk for highly relevant COVID-19 comorbid conditions may be critical to our ability to understand and therefore address the observed health disparities in the COVID-19 pandemic affecting US NHBs.

Conceptualization of multifactorial risk for COVID-19. As with any complex disease, it is the aggregate result of many risk factors and exposures which determines one’s susceptibility to and manifestation of disease. Genetic risk is likely just one piece of the complicated landscape underlying the observed disparity in Black mortality rates. Here, we highlight three candidate genes (angiotensin converting enzyme 2, ACE2; acetylcholinesterase, AChE; interleukin-6, IL-6) based on their involvement in both environmental responses and comorbid conditions that are relevant to SARS-CoV-2 pathophysiology. Importantly, it is the interplay between key environmental exposures (stress; social determinants of health, SDH) and genetic predisposition for aspects of viral pathogenesis and/or comorbid disease (e.g., type 2 diabetes mellitus, T2DM; hypertension, HTN) that ultimately converges on COVID-19 manifestation and affects mortality
Medical Comorbidities: a Major Role for the Disproportionate Infectivity and Mortality Among NHBs with SARS-CoV-2
Throughout the world and in the USA, pre-existing health risks demonstrate poorer clinical outcomes from SARS-CoV-2 [14, 15]. In the USA, NHBs have among the highest comorbidity rates of obesity, cardiovascular, hypertension, kidney, T2DM, and respiratory -related disease [16,17,18,19]. Since the SARS-CoV-2 outbreak in Wuhan, China, studies are revealing patient risk factors of mortality. In a study of 191 patients from the Wuhan Pulmonary Hospital, it was shown that 48% of patients had pre-existing chronic disease including HTN, coronary heart disease, and T2D [20]. In the USA, epidemiological data from Centers for Disease Control and Prevention shows that the risk of death from SARS-CoV-2 is significantly correlated with individuals having underlying chronic diseases/conditions, with particular devastation and mortality brought against the aged and those with underlying cardiovascular disease (CVD), HTN, and T2DM, among others [20,21,22,23,24,25,26,27,28]. NHBs have poorer overall cardiovascular health than non-Hispanic Whites, and cardiovascular disease mortality is higher in NHBs than Whites [29, 30]. Co-risk factors such as HTN, peripheral arterial disease, and T2DM have been shown to be twice as high in NHBs compared with Whites, even when controlling for risk behaviors (e.g., tobacco use, alcohol consumption) [31]. Specifically, HTN prevalence in NHBs has been reported to be as high as 57.6% for males and 53.2% for females, rates that are among the highest in the world [30, 32]. As a result, and not surprisingly, NHBs are significantly at higher risk for complications from SART-CoV-2 infection.
In a recent report by Price-Harwood et al., Black COVID-19 patients in Louisiana’s Ocher Clinic Health System, which represented 76.9% of the hospitalized, had higher prevalence of comorbid disease such as obesity, T2DM, HTN, and chronic kidney disease (Price-Haywood EG, Burton J, Fort D, Seoane L. Hospitalization and Mortality among Black Patients and White Patients with Covid-19. N Engl J Med. 2020;382(26):2534–2543. doi:https://doi.org/10.1056/NEJMsa2011686). Similar risks were found among Blacks in metropolitan Detroit and Georgia in which the majority had one or more existing comorbidities (Suleyman G, Fadel RA, Malette KM, et al. Clinical Characteristics and Morbidity Associated With Coronavirus Disease 2019 in a Series of Patients in Metropolitan Detroit. JAMA Netw Open. 2020;3(6):e2012270. Published 2020 Jun 1. doi:https://doi.org/10.1001/jamanetworkopen.2020.12270; Gold JAW, Wong KK, Szablewski CM, et al. Characteristics and Clinical Outcomes of Adult Patients Hospitalized with COVID-19 - Georgia, March 2020. MMWR Morb Mortal Wkly Rep. 2020;69(18):545–550. Published 2020 May 8. doi:https://doi.org/10.15585/mmwr.mm6918e1). These findings underscore the persistent associations between pre-existing health and clinical outcomes and the importance to understanding why NHBs are disproportionately at risk and die from COVID-19.
The Biology and Pathogenesis of SARS-CoV-2
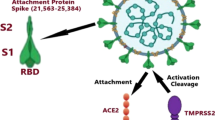
Infection of SARS-CoV-2 into cells is initiated by interaction between SARS-CoV-2 spike (S) protein and the receptor molecule, angiotensin converting enzyme 2 (ACE2), expressed on the surface of the alveolar epithelial cells [33,34,35,36,37]. The affinity of SARS-CoV-2 S protein to ACE2 and the level of ACE2 on the target cell surface are critical for virus entry and hence SARS-CoV-2 pathogenicity. In fact, recent cryo-EM structural imaging of the virus S protein showed that affinity of SARS-CoV-2 to ACE2 is 10 to 20 fold higher than that of the previous SARS-CoV [34, 38, 39], providing one explanation of how SARS-CoV-2 is more pathogenic than SARS-CoV. In addition, SARS-CoV-2 S protein contains a furin-like cleavage site not present in SARS-CoV [40], and the cellular serine protease transmembrane protease serine 2 (TMPRSS2) processes the SARS-CoV-2 S protein properly [41,42,43] to facilitate SARS-CoV-2 entry. These cumulative structural differences in the S protein of SARS-CoV-2 could be responsible for the explosiveness of morbidity and mortality in COVID-19.
We focus on the molecular function of ACE2, which plays an essential role in regulation of both underlying disease and SARS-CoV-2 entry. ACE2 is essential for metabolic control of respiratory, vascular, myocardial, kidney, and pancreatic functioning among others [44,45,46]. In metabolic disorders for example, it is known that there is an upregulation of ACE2 that interacts with the angiotensin II type 1 receptor (AT1R) which in doing so has pathologic pro-inflammatory and pro-fibrotic effects [47, 48]. Robust replication of the entered SARS-CoV-2 through ACE2 receptors triggers release of large numbers of cytokines, especially IL-6, causing acute fatal “cytokine storms”; that is, efficient entry followed by replication of the virus is essential for SARS-CoV-2 pathobiology.
ACE2 reduces angiotensin II (ANGII), the major effector of the renin-angiotensin-aldosterone system that promotes HTN, by converting ANGII to Ang-(1–7) which increases blood pressure. Interaction of the S1 domain of the S protein of SARS-CoV-2—a cleavage product of S via the host protease TMPRSS2 at the time of entry—with ACE2 induces internalization of ACE2 [34, 49, 50], resulting in loss of ACE2 on the surface of the target cells so as to increase the overall ratio of ANGII/Ang-(1–7). SARS-CoV-2 infection-mediated reduction of ACE2 thereby increases ANGII and may attribute to exacerbation of pulmonary injury and consequent tissue fibrosis [50]. In addition, the level of ACE2 expression is lowered in patients with T2DM, probably due to glycosylation, and thus the conversion of ANGII to Ang-(1–7) is prohibited, again increasing the AGNII/Ang-(1–7) ratio, which might explain the increased predisposition to severe lung injury and acute respiratory distress syndrome (ARDS) [51, 52]. Taken together, these reports demonstrate that ACE2 is a prominent mediator as well as regulator for SARS-CoV-2 infection and underlying diseases, although molecular mechanisms for how SARS-CoV-2 infection exerts such deleterious clinical impacts on patients with underlying diseases are unidentified.
COVID-19 Susceptibility: Genetic Polymorphisms in ACE2 as a Source of Biological Risk
It is known that SDH and environmental factors drive observed disparate mortalities between patients with and without underlying diseases; however, to a lesser extent, complementary biological mechanisms derived from ACE2 polymorphism-mediated elevation of SARS-CoV-2 entry may contribute to the disease. According to the National Center for Biotechnology Information (NCBI) records, most of the ACE2 polymorphisms are single nucleotide polymorphisms (SNPs), and only 34 out of 160 ACE gene polymorphisms are located in coding regions [53]. Even 18 of the 34 ACE2 coding region polymorphisms are missense mutations, establishing that most of the polymorphisms are non-functional [53]. Yet a recent report shows that many human ACE2 variants, such as S19P, I21V, and E23K, alter SARS-CoV-2-host interaction and thereby increase potential host susceptibility, and other mutants of ACE2, such as K31R, N33I, and H34R, are predicted to reduce binding activity of ACE2 to SARS-CoV-2 S protein [54, 55], indicating that ACE2 polymorphisms are likely to play a vital role in modulation of SARS-CoV-2 susceptibility.
It is also reported that treatment of HTN with ACE inhibitors and angiotensin II type 1 receptor blockers upregulates expression of ACE2 [56] and the expression of ACE2 is substantially enhanced in patients with T2DM, who are treated with ACE2 inhibitors and ATR1 receptor blockers [57,58,59], intimating that promoter polymorphisms are elemental to regulation of ACE2 expression. Collectively, these reports indicate that gene and promoter polymorphisms of ACE2 are integral to regulating SARS-CoV-2 susceptibility and ACE2 levels on the surface of target cells, respectively.
COVID-19 Pathogenesis: Genetic Polymorphisms in Acetylcholinesterase (AChE) and IL-6-Related Psychosocial Stress and Chronic Inflammatory Disease
Psychosocial stress has been associated as a risk factor of cardiovascular and other disease conditions [60], including those related to differences in the impact of stress between racial/ethnic groups [61]. Given that the origins of an individuals’ stress is multi-factorial (e.g. trauma, interpersonal, environmental), it is likely to incite broad and complex physiological responses. For example, studies link multiple domains of psychosocial stress to result in abnormalities in neuro-hormonal, central, and autonomic and immune/inflammatory systems [62, 63]. To date, it is still unclear whether specific stressors or cumulative stress and CVD risks vary based on race/ethnicity [61]. Moreover, whether or not such forms of psychosocial stress may be plausible mechanisms by which SARS-CoV-2 susceptibility is linked to racial/ethnic disparities in cardiovascular risk remain unknown.
In addition to the aforementioned implications of ACE2 in regulating SARS-CoV-2 susceptibility, other integral polymorphisms of cellular molecules with respect to SARS-CoV-2-associated pathogenicity are AChE and IL-6, a crucial regulator of blood pressure and a culprit in SARS-CoV-2-associated cytokine storm, respectively. Previous studies demonstrate the influences of stress on increased biomarkers, blood pressure control, and endothelial dysfunction [64,65,66]. AChE participates in regulation of HTN by regulating the availability of acetylcholine (ACh) which is vital in regulating blood pressure as well as anxiety/stress [67,68,69], a factor in significant underlying disease closely associated with SARS-CoV-2 mortality. SARS-CoV-2 infection causes mild to severe cytokine storm in patients, and IL-6 plays a major role in cytokine release syndrome (CRS) [70,71,72,73,74,75]. Since the IL-6 level is upregulated as the severity of COVID-19 increases, IL-6 is regarded prognostically, as a chief pathobiologic indicator for survival of symptomatic COVID-19 patients [20, 76,77,78].
MicroRNAs (miRNAs) are a class of non-coding ribonucleic acids (RNAs) that play important roles in regulating gene expression. Interestingly, the level of AChE, known to be targeted by microRNA-132 (miR132) [79], is regulated by miR608, which also modulates expression of IL-6 [80]. That is, miR608 simultaneously interacts with both targets, and regulation of these interactions is determined by the genetic polymorphisms at the 3′ untranslated region (3′ UTR) of AChE message and the promoter polymorphisms of IL-6 [80]. Specifically, miR608 suppresses expression of AChE by interacting with the 3′ UTR of AChE message, which in turn reduces availability of Ach, lowering blood pressure, while enhancing anxiety/stress [80]. However, once the AChE allele has an SNP in the 3′ UTR, the interaction between AChE and miR608 is weakened, which potentiates miR608-mediated suppression of other targets, in this case IL-6 [80]. In this manner, miR608 may participate in regulation of both blood pressure and cytokine storm in SARS-CoV-2 infected cells. However, just how miR608 suppresses IL-6, even if IL-6 also harbors both gene [81, 82] and promoter polymorphism [83], is poorly understood.
Ancestral Variation in ACE2, AChE, and IL-6 and Previously Reported Clinical Associations
Ancestral variations in ACE2, AChE, and IL-6 have previously reported clinical associations. Population-specific risk in Black communities is clearly multifactorial; however, recent research on the prevalence and risk in the UK indicates that comorbidity and social determinants of health only tell part of the story when it comes to accounting for disease risk and mortality in vulnerable populations [84]. Naturally, biological risk likely fills the void, at least in some part, and recent conversations have invoked DNA variation in a variety of pathways as possible contributors; for example, variants affecting vitamin status, coagulation factors, and immunoglobulin variation (among others) have been proposed (see opinion pieces by Valerie Palmer and Kevin Davies, accompanying Wise 2020). Here, we turn our attention to altered regulation of the immune response, which we have learned through comparative studies has a distinct evolutionary history when comparing individuals of African to European ancestry [85]. Quach et al. demonstrated that the DNA variants which exhibit the greatest divergence between African and European ancestry are in genes/regions responsible for regulating the immune system’s response specifically to viral insults. Therefore, we posit that genetic variation in stress and/or immune response confers risk by altering viral susceptibility and/or viral pathophysiology. Of particular importance as it relates to minority population health is the fact that regulation of gene expression is a process under strong epigenetic control and, therefore, is likely to change in response to environmental and psychosocial exposures. The combined effects of genetic variants in the promoters and non-coding regions of this gene set are of interest because they [86] (1) have differential allele frequencies based on ancestral populations; (2) have been previously associated with risk for HTN, T2DM, and cardiovascular susceptibilities; and (3) are relevant to COVID-19 pathophysiology (see Supplementary Table 1).
ACE2 SNPs previously implicated in HTN largely based on studies of European and Chinese populations are known to have elevated allele frequencies in African ancestral populations. Although at this time, the significance of these variations in ACE2 with respect to HTN are controversial [87,88,89,90,91,92], it is intriguing to note that the variants in ACE2 with the highest discrepancy in allele frequency between African and European ancestry are also the variants uncovered in multiple studies of relevant COVID-19 comorbidity risks (see Supplementary Table 1). Similarly, the key SNP in the 3′ UTR region of the AChE gene, rs17228616, which modulates miRNA targeting and off-target effects as a result, is far more common in populations of African ancestry when compared with European. Given the effect of this SNP on IL-6 production, it follows that this genetic variant may alter the progression and severity of SARS-CoV-2 response and, likewise, that variants in IL-6 may also impact miRNA alterations to gene expression. Indeed, many studies of IL-6 haplotype effects have reported significant associations with cardiovascular function as well as metabolic disease (i.e., T2DM). While there is conflicting data regarding the effects of variants in all three of the candidate genes discussed here, the remarkable relevance of associated phenotypes to COVID-19 pathophysiology together implies that genetic polymorphisms which regulate immune and stress responses may interact to affect underlying disease risk and, simultaneously, SARS-CoV-2 pathogenicity.
Concluding Remarks
Persistent, low-level inflammation is a prevailing hallmark of most if not all chronic disease and considered responsible for health outcomes [93,94,95]. Juxtaposed with the robust cytokine-mediated disease etiology and underlying biopsychosocial factors demarcates the setting of a “perfect storm.” The accumulated works presented here merge the multi-faceted influences of comorbid risk, the emerging biomolecular etiology of CoV-2 pathogenesis, and the underlying preferences in heritable genetic predisposition, as a collective explanation of the observed disparities of COVID-19 deaths in NHBs.
Clearly, there are many complicated interactions between biological and environmental sources of risk for severe COVID-19 outcomes; large-scale research of vulnerable human populations is warranted to enable the identification and quantification of the key drivers for COVID-19 mortality. Studies of this nature may allow for the development of models which predict risk at the individual level (Fig. 2), where the nature of exposures (e.g., SDH, psychosocial stress) can be weighted alongside the effects of specific genetic risk loci (e.g., where A, B, and C may be variants in genes such as ACE2, AChE, and IL-6) in order to determine the likelihood and/or degree of adverse COVID-19 pathology upon exposure. While this theoretical “threshold” model of disease risk is a vast oversimplification, rapid advances in computational biology, facilitated by artificial intelligence, may enable researchers to capture nuanced interactions not shown in Fig. 2 in order to greatly enhance predictive power.

Theoretical “threshold” model for COVID-19. Visualization of how multifactorial risk factors may theoretically combine to inform predictive models of disease risk at the individual level
To conclude, ongoing research in the field of health disparities provides evidence that the persistent disproportion of disease for NHBs and other ethnic/racial minorities and underserved groups will not be eliminated without concerted efforts to address social determinants. It is also true that a conscientious view into biologically derived risk and its interconnections within the psychosocial and environmental realms is required to realize the full potential of the knowledge gained to stimulate progress in therapeutics for decreasing health disparities in NHBs and conquering this pandemic as well as improve health outcomes.
Data Availability
Not applicable.
References
Wu LT, Blazer DG. Substance use disorders and co-morbidities among Asian Americans and Native Hawaiians/Pacific Islanders. Psychol Med. 2015;45(3):481–94.
Kim EJ, Kressin NR, Paasche-Orlow MK, Lopez L, Rosen JE, Lin M, et al. Racial/ethnic disparities among Asian Americans in inpatient acute myocardial infarction mortality in the United States. BMC Health Serv Res. 2018;18(1):370.
Cyrus E, Clarke R, Hadley D, Bursac Z, Trepka MJ, Devieux JG, et al. The impact of COVID-19 on African American communities in the United States. medRxiv. 2020.
Hussain A, Boulton AJM. COVID-19 and diabetes: International diabetes federation perspectives. Diabetes Res Clin Pract. 2020;167:108339.
Palaiodimos L, Kokkinidis DG, Li W, Karamanis D, Ognibene J, Arora S, et al. Severe obesity, increasing age and male sex are independently associated with worse in-hospital outcomes, and higher in-hospital mortality, in a cohort of patients with COVID-19 in the Bronx, New York. Metabolism. 2020;108:154262.
Yancy CW. COVID-19 and African Americans. Jama. 2020;323:1891–2.
Baum A, Garofalo JP, Yali AM. Socioeconomic status and chronic stress. Does stress account for SES effects on health? Ann N Y Acad Sci. 1999;896:131–44.
Gough M, Godde K. A multifaceted analysis of social stressors and chronic inflammation. SSM Popul Health. 2018;6:136–40.
Aschbacher K, O'Donovan A, Wolkowitz OM, Dhabhar FS, Su Y, Epel E. Good stress, bad stress and oxidative stress: insights from anticipatory cortisol reactivity. Psychoneuroendocrinology. 2013;38(9):1698–708.
Coppack SW. Pro-inflammatory cytokines and adipose tissue. Proc Nutr Soc. 2001;60(3):349–56.
Bertholdt L, Gudiksen A, Jessen H, Pilegaard H. Impact of skeletal muscle IL-6 on regulation of liver and adipose tissue metabolism during fasting. Pflugers Arch. 2018;470(11):1597–613.
Hoffmann A, Ebert T, Kloting N, Kolb M, Gericke M, Jeromin F, et al. Leptin decreases circulating inflammatory IL-6 and MCP-1 in mice. Biofactors. 2019;45(1):43–8.
Zilbermint M, Hannah-Shmouni F, Stratakis CA. Genetics of hypertension in African Americans and others of African descent. Int J Mol Sci. 2019;20(5):1081.
Garg S. Hospitalization rates and characteristics of patients hospitalized with laboratory-confirmed coronavirus disease 2019—COVID-NET, 14 states, march 1–30, 2020. MMWR Morb Mortal Wkly Rep. 2020;69:458–64.
Sardu C, Gambardella J, Morelli MB, Wang X, Marfella R, Santulli G. Hypertension, thrombosis, kidney failure, and diabetes: is COVID-19 an endothelial disease? A comprehensive evaluation of clinical and basic evidence. J Clin Med. 2020;9(5):1417.
Gawlik KS, Melnyk BM, Tan A. Associations between stress and cardiovascular disease risk factors among million hearts priority populations. Am J Health Promot. 2019;33(7):1063–6.
Li L, Brennan KJ, Gaughan JP, Ciccolella DE, Kuzma AM, Criner GJ. African Americans and men with severe COPD have a high prevalence of osteoporosis. COPD. 2008;5(5):291–7.
Global Burden of Cardiovascular Diseases C, Roth GA, Johnson CO, Abate KH, Abd-Allah F, Ahmed M, et al. The burden of cardiovascular diseases among US States, 1990–2016. JAMA Cardiol. 2018;3(5):375–89.
Suleyman G, Fadel RA, Malette KM, Hammond C, Abdulla H, Entz A, et al. Clinical characteristics and morbidity associated with coronavirus disease 2019 in a series of patients in metropolitan Detroit. JAMA Netw Open. 2020;3(6):e2012270.
Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054–62.
Arentz M, Yim E, Klaff L, Lokhandwala S, Riedo FX, Chong M, et al. Characteristics and outcomes of 21 critically ill patients with COVID-19 in Washington state. Jama. 2020;323(16):1612–4.
Chiolero A. Covid-19: a digital epidemic. BMJ. 2020;368:m764
Del Rio C, Malani PN. COVID-19—new insights on a rapidly changing epidemic. JAMA. 2020;323(14):1339–40.
Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020;5:811–8.
Liu F, Xu A, Zhang Y, Xuan W, Yan T, Pan K, et al. Patients of COVID-19 may benefit from sustained lopinavir-combined regimen and the increase of eosinophil may predict the outcome of COVID-19 progression. Int J Infect Dis. 2020;95:183–91.
Mehra MR, Desai SS, Kuy S, Henry TD, Patel AN. Cardiovascular disease, drug therapy, and mortality in COVID-19. N Engl J Med. 2020;382:2582.
Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020;46(5):846–8.
Yang J, Zheng Y, Gou X, Pu K, Chen Z, Guo Q, et al. Prevalence of comorbidities in the novel Wuhan coronavirus (COVID-19) infection: a systematic review and meta-analysis. Int J Infect Dis. 2020;94:91–5.
Rosamond W, Johnson A, Bennett P, O'Brien E, Mettam L, Jones S, et al. Monitoring and improving acute stroke care: the North Carolina stroke care collaborative. N C Med J. 2012;73(6):494–8.
Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, et al. Heart disease and stroke statistics-2017 update: a report from the American Heart Association. Circulation. 2017;135(10):e146–603.
Allison MA, Gonzalez F 2nd, Raij L, Kaplan R, Ostfeld RJ, Pattany MS, et al. Cuban Americans have the highest rates of peripheral arterial disease in diverse Hispanic/Latino communities. J Vasc Surg. 2015;62(3):665–72.
Maraboto C, Ferdinand KC. Update on hypertension in African-Americans. Prog Cardiovasc Dis. 2020;63(1):33–9.
Albini A, Di Guardo G, Noonan DM, Lombardo M. The SARS-CoV-2 receptor, ACE-2, is expressed on many different cell types: implications for ACE-inhibitor- and angiotensin II receptor blocker-based cardiovascular therapies. Intern Emerg Med. 2020:1–2. https://doi.org/10.1007/s11739-020-02436-7.
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280.e8.
Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215–20.
Lukassen S, Chua RL, Trefzer T, Kahn NC, Schneider MA, Muley T, et al. SARS-CoV-2 receptor ACE 2 and TMPRSS 2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020;39(10):e105114.
Wang Q, Zhang Y, Wu L, Niu S, Song C, Zhang Z, et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell. 2020;d181(4):894–904.e9.
Chen Y, Guo Y, Pan Y, Zhao ZJ. Structure analysis of the receptor binding of 2019-nCoV. Biochem Biophys Res Commun. 2020;525(1):135–40.
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, Abiona O, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–3.
Coutard B, Valle C, de Lamballerie X, Canard B, Seidah N, Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir Res. 2020;176:104742.
Belouzard S, Chu VC, Whittaker GR. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc Natl Acad Sci. 2009;106(14):5871–6.
Glowacka I, Bertram S, Müller MA, Allen P, Soilleux E, Pfefferle S, et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J Virol. 2011;85(9):4122–34.
Shulla A, Heald-Sargent T, Subramanya G, Zhao J, Perlman S, Gallagher T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J Virol. 2011;85(2):873–82.
Chhabra KH, Xia H, Pedersen KB, Speth RC, Lazartigues E. Pancreatic angiotensin-converting enzyme 2 improves glycemia in angiotensin II-infused mice. Am J Physiol Endocrinol Metab. 2013;304(8):E874–E84.
Jia H. Pulmonary angiotensin-converting enzyme 2 (ACE2) and inflammatory lung disease. Shock. 2016;46(3):239–48.
Anguiano L, Riera M, Pascual J, Soler M. Circulating ACE2 in cardiovascular and kidney diseases. Curr Med Chem. 2017;24(30):3231–41.
Dalan R, Bornstein SR, El-Armouche A, Rodionov RN, Markov A, Wielockx B, et al. The ACE-2 in COVID-19: foe or friend? Horm Metab Res. 2020;52(5):257–63.
Shekhar S, Wurth R, Kamilaris CDC, Eisenhofer G, Barrera FJ, Hajdenberg M, et al. Endocrine conditions and COVID-19. Horm Metab Res. 2020;52(7): 471–84.
Iwata-Yoshikawa N, Okamura T, Shimizu Y, Hasegawa H, Takeda M, Nagata N. TMPRSS2 contributes to virus spread and immunopathology in the airways of murine models after coronavirus infection. J Virol. 2019;93(6):e01815-18.
South AM, Diz DI, Chappell MC. COVID-19, ACE2, and the cardiovascular consequences. Am J Phys Heart Circ Phys. 2020;318:H1084–90.
Tikellis C, Thomas M. Angiotensin-converting enzyme 2 (ACE2) is a key modulator of the renin angiotensin system in health and disease. Int J Pept. 2012;2012:1–8.
Wu C, Chen X, Cai Y, Zhou X, Xu S, Huang H, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 2020;180:934–43.
Sayed-Tabatabaei F, Oostra B, Isaacs A, Van Duijn C, Witteman J. ACE polymorphisms. Circ Res. 2006;98(9):1123–33.
Calcagnile M, Forgez P, Iannelli A, Bucci C, Alifano M, Alifano P. ACE2 polymorphisms and individual susceptibility to SARS-CoV-2 infection: insights from an in silico study. bioRxiv. 2020.
Stawiski EW, Diwanji D, Suryamohan K, Gupta R, Fellouse FA, Sathirapongsasuti F, et al. Human ACE2 receptor polymorphisms predict SARS-CoV-2 susceptibility. BioRxiv. 2020.
Li XC, Zhang J, Zhuo JL. The vasoprotective axes of the renin-angiotensin system: physiological relevance and therapeutic implications in cardiovascular, hypertensive and kidney diseases. Pharmacol Res. 2017;125:21–38.
Fang L, Karakiulakis G, Roth M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir Med. 2020;8(4):e21.
Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol. 2020;94(7):e00127–20.
Wan Y, Shang J, Sun S, Tai W, Chen J, Geng Q, et al. Molecular mechanism for antibody-dependent enhancement of coronavirus entry. J Virol. 2020;94(5):e02015–19.
Rosengren A, Hawken S, Ounpuu S, Sliwa K, Zubaid M, Almahmeed WA, et al. Association of psychosocial risk factors with risk of acute myocardial infarction in 11119 cases and 13648 controls from 52 countries (the INTERHEART study): case-control study. Lancet. 2004;364(9438):953–62.
Sternthal MJ, Slopen N, Williams DR. Racial disparities in health: How much does stress really matter? Du Bois Rev. 2011;8(1):95–113.
McEwen BS. Protective and damaging effects of stress mediators. N Engl J Med. 1998;338(3):171–9.
Friedman EM, Williams DR, Singer BH, Ryff CD. Chronic discrimination predicts higher circulating levels of E-selectin in a national sample: the MIDUS study. Brain Behav Immun. 2009;23(5):684–92.
Hatch SL, Dohrenwend BP. Distribution of traumatic and other stressful life events by race/ethnicity, gender, SES and age: a review of the research. Am J Community Psychol. 2007;40(3–4):313–32.
Poon I, Lal LS, Ford ME, Braun UK. Racial/ethnic differences in blood pressure control and medication utilization in a cohort of older veterans with dementia. Am J Ther. 2010;17(1):34–41.
Rodriguez CJ, Gwathmey TM, Jin Z, Schwartz J, Beech BM, Sacco RL, et al. Perceived discrimination and nocturnal blood pressure dipping among Hispanics: the influence of social support and race. Psychosom Med. 2016;78(7):841–50.
Handa T, Katare RG, Kakinuma Y, Arikawa M, Ando M, Sasaguri S, et al. Anti-Alzheimer's drug, donepezil, markedly improves long-term survival after chronic heart failure in mice. J Card Fail. 2009;15(9):805–11.
Inanaga K, Ichiki T, Miyazaki R, Takeda K, Hashimoto T, Matsuura H, et al. Acetylcholinesterase inhibitors attenuate atherogenesis in apolipoprotein E-knockout mice. Atherosclerosis. 2010;213(1):52–8.
Lataro RM, Silva CA, Tefé-Silva C, Prado CM, Salgado HC. Acetylcholinesterase inhibition attenuates the development of hypertension and inflammation in spontaneously hypertensive rats. Am J Hypertens. 2015;28(10):1201–8.
Miossec P, Kolls JK. Targeting IL-17 and TH 17 cells in chronic inflammation. Nat Rev Drug Discov. 2012;11(10):763–76.
Chousterman BG, Swirski FK, Weber GF. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol: Springer. 2017;39:517–28.
Teijaro JR. Cytokine storms in infectious diseases. Semin Immunopathol: Springer. 2017;39:501–3.
Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24(6):739–48.
Shimabukuro-Vornhagen A, Gödel P, Subklewe M, Stemmler HJ, Schlößer HA, Schlaak M, et al. Cytokine release syndrome. J Immunother Cancer. 2018;6(1):56.
Liu B, Li M, Zhou Z, Guan X, Xiang Y. Can we use interleukin-6 (IL-6) blockade for coronavirus disease 2019 (COVID-19)-induced cytokine release syndrome (CRS)? J Autoimmun. 2020;111:102452.
Chen L, Liu H, Liu W, Liu J, Liu K, Shang J, et al. Analysis of clinical features of 29 patients with 2019 novel coronavirus pneumonia. Zhonghua Jie He He Hu Xi Za Zhi. 2020;43:E005–E05.
Gao Y, Li T, Han M, Li X, Wu D, Xu Y, et al. Diagnostic utility of clinical laboratory data determinations for patients with the severe COVID-19. J Med Virol. 2020;92:791–6.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506.
Shaked I, Meerson A, Wolf Y, Avni R, Greenberg D, Gilboa-Geffen A, et al. MicroRNA-132 potentiates cholinergic anti-inflammatory signaling by targeting acetylcholinesterase. Immunity. 2009;31(6):965–73.
Hanin G, Shenhar-Tsarfaty S, Yayon N, Yau YH, Bennett ER, Sklan EH, et al. Competing targets of microRNA-608 affect anxiety and hypertension. Hum Mol Genet. 2014;23(17):4569–80.
Fishman D, Faulds G, Jeffery R, Mohamed-Ali V, Yudkin JS, Humphries S, et al. The effect of novel polymorphisms in the interleukin-6 (IL-6) gene on IL-6 transcription and plasma IL-6 levels, and an association with systemic-onset juvenile chronic arthritis. J Clin Invest. 1998;102(7):1369–76.
Talar-Wojnarowska R, Gasiorowska A, Smolarz B, Romanowicz-Makowska H, Kulig A, Malecka-Panas E. Clinical significance of interleukin-6 (IL-6) gene polymorphism and IL-6 serum level in pancreatic adenocarcinoma and chronic pancreatitis. Dig Dis Sci. 2009;54(3):683–9.
Giacconi R, Cipriano C, Albanese F, Boccoli G, Saba V, Olivieri F, et al. The− 174G/C polymorphism of IL-6 is useful to screen old subjects at risk for atherosclerosis or to reach successful ageing. Exp Gerontol. 2004;39(4):621–8.
Wise J. Covid-19: known risk factors fail to explain the increased risk of death among people from ethnic minorities. London: British Medical Journal Publishing Group; 2020;369:m1873.
Quach H, Rotival M, Pothlichet J, Loh Y-HE, Dannemann M, Zidane N, et al. Genetic adaptation and neandertal admixture shaped the immune system of human populations. Cell. 2016;167(3):643–56 e17.
Fitzpatrick L. Scarcity in digital health innovation for Medicaid populations: is help on the way? 2019.
Doris PA. Hypertension genetics, single nucleotide polymorphisms, and the common disease: common variant hypothesis. Hypertension. 2002;39(2):323–31.
Benjafield AV, Wang WY, Morris BJ. No association of angiotensin-converting enzyme 2 gene (ACE2) polymorphisms with essential hypertension. Am J Hypertens. 2004;17(7):624–8.
Lieb W, Graf J, Götz A, König IR, Mayer B, Fischer M, et al. Association of angiotensin-converting enzyme 2 (ACE2) gene polymorphisms with parameters of left ventricular hypertrophy in men. J Mol Med. 2006;84(1):88–96.
Zhong J, Yan Z, Liu D, Ni Y, Zhao Z, Zhu S, et al. Association of angiotensin-converting enzyme 2 gene A/G polymorphism and elevated blood pressure in Chinese patients with metabolic syndrome. J Lab Clin Med. 2006;147(2):91–5.
Pan Y, Wang T, Li Y, Guan T, Lai Y, Shen Y, et al. Association of ACE2 polymorphisms with susceptibility to essential hypertension and dyslipidemia in Xinjiang, China. Lipids Health Dis. 2018;17(1):241.
Luo Y, Liu C, Guan T, Li Y, Lai Y, Li F, et al. Association of ACE2 genetic polymorphisms with hypertension-related target organ damages in south Xinjiang. Hypertens Res. 2019;42(5):681–9.
Shields GS, Moons WG, Slavich GM. Inflammation, self-regulation, and health: an immunologic model of self-regulatory failure. Perspect Psychol Sci. 2017;12(4):588–612.
Lin C, Michopoulos V, Powers A, Wingo AP, Schwartz A, Bradley B, et al. Affect, inflammation, and health in urban at-risk civilians. J Psychiatr Res. 2018;104:24–31.
Raggi P, Genest J, Giles JT, Rayner KJ, Dwivedi G, Beanlands RS, et al. Role of inflammation in the pathogenesis of atherosclerosis and therapeutic interventions. Atherosclerosis. 2018;276:98–108.
Funding
Research reported in this publication was supported by the National Institute on Minority Health and Health Disparities of the National Institutes of Health under Award Number U54MD006882.
Author information
Authors and Affiliations
Contributions
All authors (NRP, IP, JR, HJ) contributed to content curation, writing, and review of this manuscript. HJ was responsible for oversight, coordination, and conceptualization; NRP was responsible for coordination, generating figures/tables, and formatting for submission.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Code Availability
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(DOCX 424 kb).
Rights and permissions
About this article

Cite this article
Phillips, N., Park, IW., Robinson, J.R. et al. The Perfect Storm: COVID-19 Health Disparities in US Blacks. J. Racial and Ethnic Health Disparities 8, 1153–1160 (2021). https://doi.org/10.1007/s40615-020-00871-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40615-020-00871-y