
Abstract
To investigate its application as a material for solid oxide fuel cell interconnects, oxidation rates of replicated E-Brite (Fe-26Cr-1Mo, wt pct) foams with 43 and 51 pct open porosity were measured in static laboratory air for up to 200 hours. Results correlate well with previously reported values for dense material when normalized by surface area. Area-specific resistance measurements, taken in the range of 823 K to 1073 K (550 °C to 800 °C) after 24 hours of oxidation at 1123 K (850 °C), yield activation energies in the range 69 to 82 kJ mol−1 for porous E-Brite that closely match dense E-Brite. Compressive creep properties, measured at 1073 K (800 °C) for pristine and oxidized porous E-Brite, show that pre-oxidation (10 hours at 1073 K (800 °C)) led to a ~100-fold decrease in creep rate. This is due to strengthening of the alloy foam by the formation of a continuous network of oxide, which coats the internal pore surface and reduces porosity by as much as 10 pct after 200 hours of oxidation at 1073 K (800 °C). Choking of the fenestrations between the pores, however, leads to an increase in closed porosity. Strengthening and pore filling effects should be taken into account in the design of the SOFC stack when using E-Brite as a porous interconnect material.
Similar content being viewed by others


Introduction
Solid oxide fuel cells (SOFCs), which are some of the most efficient devices for converting fuel to electricity, have so far mostly been considered for use in stationary applications such as centralized power plants. Research in recent years has also highlighted SOFCs’ potential use as auxiliary power units for transportation systems, e.g., for aerospace, marine, and trucking (long-haul/idling) applications.[1–5] Service requirements are similar to those for stationary applications but with additional emphasis on ruggedness and reduced weight.[6] Traditional SOFCs have utilized anode-supported, cathode-supported, and electrolyte-supported designs, which receive their mechanical support from the anode, cathode, and electrolyte, respectively. However, the need for lightweighting and ruggedness can be addressed by implementing alternative stack designs such as metal-supported cells, where robust mechanical support is provided by a lightweight, ductile, porous metal.[4,7] The anode, electrolyte, and cathode can be deposited directly on top of metal-supported cells. This has led to replacement of expensive La-based ceramics with ferritic steels[6,8] and can reduce the thickness of yttria-stabilized zirconia used in the electrolyte. As described in previous work,[9] cells are commonly connected in stacks in which case a porous metal can be added to either side of a non-porous core thus allowing the foam to serve as a channel for fuel and oxidant while the non-porous core serves as a physical barrier.
Potential metals for use in metal-supported cells, as in the case of dense substrates, remain limited due to the harsh environmental conditions imposed by simultaneous exposure to an oxidizing and reducing atmosphere at temperatures as high as 1073 K (800 °C),[10] Consequently, ferritic stainless steels remain one of the most studied candidate families of alloys due to their coefficient of thermal expansion matching with the stack, electrically conducting oxide (chromia, Cr2O3), ease of fabrication, and low cost. Several studies have characterized the oxidation behavior, thermal expansion, and area-specific resistance (ASR) of dense ferritic steels for SOFC operating conditions.[10–12]
Porous ferritic stainless steels suitable for metal-supported cells have been successfully prepared by a variety of methods including: laser drilling,[13–15] tape-casting,[16] and prealloyed powder isostatic pressing.[7,17,18] Porosities are typically in the range of 30 to 50 pct so as to allow sufficient gas flow across the electrodes. However, the high surface area of these porous structures can lead to rapid oxidation.[19] Oxide growth and the concomitant loss of conductivity are affected by the presence of defect concentrations or minor phases. Defects play a key role in transport through the scale and previous work has reported differences in conductivity of up to two orders of magnitude for in situ measurements when compared to the bulk.[6] The oxide growth kinetics are therefore best studied in situ, but evaluation of this behavior in porous stainless steels is limited to a handful of alloys that includes: 316L,[20] 430L,[2,16] Fe22Cr,[21] and 70Fe30Cr.[19] Furthermore, only the studies by Molin et al.[2,20] on porous 316L and 430L investigated the ASR, which is used to assess service lifetimes. Both 316L and 430L exceeded the commonly cited resistance limit of 100 mΩcm2 within 200 and 60 hours, respectively, of air exposure at 1073 K (800 °C).
In addition to affecting resistivity, the presence of an oxide can also alter mechanical properties of porous metals. It was previously shown in the case of porous Fe-26Cr-1Mo that strain rates at 1123 K (850 °C) steadily decrease, at constant stress, with increased exposure to air due to the formation of oxide, which provides a strong, continuous layer that reinforces the foam.[3] Increased creep resistance may be beneficial for stack designs, as it reduces cell deformation under stresses, especially in transportation applications where shocks (e.g., during plane landing or truck driving) can be expected. Furthermore, residual stresses due to thermal expansion mismatch are often present in SOFC consisting of various materials bonded together to achieve a gas-tight arrangement. Oxide growth also has negative effects as it reduces electrical conductivity, and chokes fenestrations between pores, thereby hindering gas flow. It is thus desirable to fully understand the resistivity and mechanical properties of porous alloys for SOFC as a function of oxidation time to ensure long-term performance of the fuel cell stack. The present paper examines the electrochemical and mechanical properties with respect to oxidation for porous E-Brite, and makes comparison with fully dense E-Brite and other porous interconnect alloys.
Experimental Section
Processing
E-Brite (Fe-26Cr-1Mo, wt pct), a commercial ferritic stainless steel alloy developed by Allegheny Ludlum (Pittsburgh, PA) for use in SOFC interconnects, was chosen because of its coefficient of thermal expansion match with ceramic SOFC components, oxidation resistance, and low material cost.[10] Porous billets (~26 mm diameter × 10 mm height) were prepared by vacuum sintering at 1523 K (1250 °C) for 4 hours of a blend of Fe (APS 6 to 10 µm), Cr (APS < 10 µm), Mo (APS 3 to 7 µm) and NaCl (crushed and sieved to 53 to 106 µm) powders. The powders were mixed for 60 minutes in a polymer bottle and cold-pressed in a die to 350 MPa. Sintering led to interdiffusion of the metallic powders and complete evaporation of the 40 or 50 vol pct of NaCl space-holder, as confirmed by chemical analysis of the sintered billets. Further details concerning the fabrication process are given in Reference 3. The extent of closed porosity in as-sintered billets was determined by helium pycnometry, using a value of 7.69 g cm−3 for dense E-Brite.[3] Measurements of the total billet porosity and individual sample porosity were performed by the Archimedes method in water, after coating the porous samples with a thin layer of vacuum grease to prevent ingress of water into the open porosity. When reporting porosity, this total porosity is used, unless otherwise specified.
One billet was prepared as described above but with no NaCl place-holder. The sintered compact was subsequently melted at 1723 K (1450 °C) in a furnace with 0.08 MPa Ar atmosphere to achieve a full density control sample.
Oxidation Measurements
Cylindrical specimens with 6.0 mm diameter and 12.0 mm height were cut by electro-discharge machining from the billets. These specimens were placed in an alumina boat and cyclically oxidized in static laboratory air (uncontrolled humidity) in a muffle furnace at 1073 K (800 °C) for up to 250 hours. Samples were periodically returned to ambient temperature to allow measurement of weight gain from oxidation. The weight increase was normalized by the surface area, which was determined using dedicated specimens as follows. Foams with 43.2 and 51.4 pct porosity (labeled in the following as 43 and 51 pct) were simultaneously oxidized for ~5 hours with an E-Brite plate (~1.5 g mass) with an easily determinable surface area. Under the assumption that surface area was proportional to weight gain for each sample, the ratio of the plate’s surface area to weight gain was used to find each foam’s surface area.
Creep Measurements
Three cylindrical specimens (6 mm diameter × 12 mm height) were cut from each billet to determine the creep response under argon after 0, 10, and 100 hours of prior exposure to laboratory air at 1073 K (800 °C). Constant-load creep tests were performed in compression using an ATS Series 3210 creep frame with a lever arm that magnified the load by a factor of three. Sample strain was measured by a linear variable differential transducer that monitored deflection of the alumina pushrod to a precision of 6 µm. A testing temperature of 1073 K (800 °C), chosen to simulate typical SOFC operating conditions, was achieved by means of a three-zone, resistively heated furnace with a temperature stability of ±2 °C. A retort setup was assembled inside the furnace using a quartz tube to enable flowing argon and insure negligible oxidation throughout the test, as confirmed by mass measurements before and after testing: weight gain was <1 mg (~0.07 pct) for a testing time of up to 3 days. Up to four increasing loads were applied on a single sample, but in all cases the accumulated compressive strain did not exceed 10 pct. Corresponding secondary creep rates were determined by the slopes of strain–time plots acquired for each stress, ignoring the initial primary creep region.
Resistivity Measurements
ASR measurements were performed on foams with both porosities and on the fully dense control sample using a four-probe setup shown schematically in Figure 1. All samples were cut with a low-speed diamond saw to a thickness of approximately 2 (±0.5) mm and an area of 10 × 10 (±1) mm2, with the actual sample area measured precisely and used in the ASR calculation. The samples were polished on both faces to 1200 grit SiC paper and subsequently pre-oxidized in laboratory air (without controlled humidity) inside a muffle furnace at 1123 K (850 °C) for 24 hours. A temperature of 1123 K (850 °C) was used for pre-oxidation because it was higher than the maximum ASR testing temperature, thus reducing any confounding effects of oxide growth during experimentation. Both surfaces of the samples were then coated with Ag paste (C4400UF, Heraeus, W. Conshohocken, PA) as electrodes and calcined in a muffle furnace at 1023 K (750 °C) in air for 15 minutes. Leads of Ag wire (0.05 mm diameter, Alfa Aesar, Ward Hill, MA) were then attached with additional Ag paste and the specimens were again placed in a muffle furnace at 1023 K (750 °C) in air for 15 minutes.

Schematic of setup used for ASR measurements of porous and dense E-Brite
Prior to high temperature testing, a DC current range of 10 to 100 mA was applied and the resulting variation in voltage was monitored in order to verify Ohmic behavior indicative of good bonding between sample and electrodes. Electrical resistance of the oxide scale was determined by heating the samples in laboratory air using a muffle furnace from 823 K to 1073 K (550 °C to 800 °C) with 50 K (50 °C) steps and cooling it at the same rate. Each temperature was held for ~30 minutes, at which time a current of 10 mA was applied and the voltage drop was recorded with a HP 34401A multimeter. Temperature was monitored with a K-type thermocouple located ~1 cm from the sample. Two measurements were taken at each temperature—one upon heating and the other upon cooling—in order to verify reproducibility.
Selected specimens were cross-sectioned, mounted in epoxy resin, and polished to 0.05 µm using colloidal alumina and standard metallographic procedures. Observations of the alloy, scale, and Ag electrode were performed with a Hitachi S-3400 N-II variable-pressure scanning electron microscope (SEM). Energy dispersive X-ray (EDX) analysis was also performed on cross-sections of ASR specimens to characterize potential electromigration of the electrode.
X-ray diffraction
X-ray diffraction (XRD) spectra were collected on two as-received E-Brite sheets from Allegheny Ludlum and two fully densified Fe-26Cr-1Mo control sample. For each pair, the face of one sample was polished to 1200 grit while the other was oxidized in air at 1123 K (850 °C) for 24 hours. Diffraction results were obtained using a Scintag XDS2000 automated diffraction system with measurements performed in air at room temperature using a Cu Kα source. The samples were scanned over the range of 2θ = 20 to 80 deg.
Results and Discussion
Oxidation Behavior
Figure 2 displays the kinetics of mass gain at 1073 K (800 °C) in static laboratory air up to 200 hours for four E-Brite foams: two with 43 pct porosity and two with 51 pct porosity. When normalized by sample original mass as in Figure 2(a), the mass gains of the 51 pct porous foams are higher than those of the 43 pct porous foams, as expected due to the difference in surface area. However, if normalized by total (inner and outer surface) area as in Figure 2(b) (and explained in Section II), differences are small and probably within experimental error, indicating that the mechanism of oxidation for the two foams are the same in the static air environment. A small amount of microcracking and subsequent oxide growth likely occurred when removing samples for mass measurement but the multi-cycle growth is still closely modeled by Wagner’s oxidation theory, as demonstrated with previous work on multi-cycle oxidation of porous metallic interconnects,[20] where scale growth is dominated by diffusion. It predicts a parabolic dependence for the mass gain per unit area Δm:
where K p is the parabolic rate constant, and t is time. It has been shown in previous work[22] that this may not be a suitable prediction for long-term behavior as K p varies over time. As postulated in that work, chromium evaporation and depletion from the oxide may be partially responsible for the observed deviation from linearity evident in Figure 3, which plots Δm 2 as a function of t. Thus, a more appropriate method of comparison is to determine the instantaneous value of K p, which was accomplished by determining the slope of every pair of neighboring data points in Figure 3. The resulting range of K p values for the samples tested is collected in Table I.

Oxidation kinetics for 43 and 51 pct porous E-Brite foams at 1073 K (800 °C) in static laboratory air, with mass gain normalized by (a) sample weight and (b) sample total (inner and outer) surface area

Oxidation kinetics for E-Brite foams at 1073 K (800 °C) in static laboratory air shown as the square of weight gain normalized by sample total surface area. (Mass gain data shown in Fig. 2)
Some mass loss is possible due to the evaporation of chromia according to the following equation:
However, airflow was minimized through the box furnace by plugging any visible holes with alumina foam, thus decreasing the likelihood of any significant effects from chromia evaporation.
As the scale grows, pore size decreases thus choking fenestrations and reducing open porosity as reported in Table II. The oxide growth within the pores and the resulting decrease in open porosity are visible in the series of optical micrographs shown in Figure 4 for cross-sections of samples oxidized for 0, 10, 100, and 200 hours in static laboratory air at 1073 K (800 °C).

Optical micrographs of polished cross-sections for 51 pct porous E-Brite foam after 0, 10, 100, and 200 h of exposure at 1073 K (800 °C) in laboratory air. Oxide (dark gray) is visible coating pore surfaces in E-Brite matrix (white) after just 10 h of exposure. Some regions of thicker oxide scales are highlighted with arrows
The range of values for parabolic oxidation rates of porous E-Brite correlates well with previously reported values for the bulk material, as shown in Table I. Among chromia-forming ferritic stainless steel alloys, this rate is low compared to potential interconnect alloy alternatives such as 430 or 446, with values of 3.5 × 10−13 and 1.3 × 10−13 g2cm−4s−1, respectively, at 1073 K (800 °C).[10] Oxide spallation, however, was visible beyond ~50 hours and remains an issue with chromia-forming alloys. Oxide cracking is also a common problem, particularly during prolonged exposure and when subject to thermal cycling.[23]
Although scale growth continues well beyond 200 hours of exposure, the non-linear behavior shown in Figure 3 may be due to the decreases in porosity induced by pore choking This is evidenced by Table II and implies that a larger initial porosity can be used to maintain a desired threshold (minimum open porosity) for sufficient flow of fuel and oxidant through the foam. The decrease in porosity stems from the reduction in pore—and in particular fenestrations—size, as illustrated in Figure 4. Given that the metal/oxide ratio was unknown, it was not possible to analyze the reduction in closed porosity using pycnometry. Usable open porosity was nonetheless determined by the volumetric difference between helium pycnometry and Archimedes measurements performed on the same samples coated with a thin layer of vacuum grease. As expected, open porosity decreases: after 200 hours of exposure, a ~10 pct reduction has accumulated.
Creep Properties
A typical compressive strain–time plot at 1073 K (800 °C) is shown in Figure 5 for a 43 pct porous foam preoxidized for 10 hours at 1073 K (800 °C) in air prior to creep testing in argon. All creep curves exhibited similar behavior that was also consistent with the power-law creep observed in previous work on porous E-Brite,[3] consisting of an initial stage of primary creep followed by a secondary stage where the average strain rate was constant. Samples were removed prior to tertiary stage creep (i.e., before densification).

Typical compressive strain–time plot of a 43 pct porous E-Brite foam, preoxidized in laboratory air for 10 h at 1073 K (800 °C), upon creep under flowing argon at 1073 K (800 °C) for four successive stresses. Stresses and secondary minimum strain rates are shown next to each experiment
As shown in Figure 6, the creep data for 43 and 51 pct porous E-Brite preoxidized for 0, 10, and 100 hours can be described by a power-law:
where A is the Dorn constant, n the stress exponent, Q the activation energy, R the gas constant, and T temperature. Best-fit apparent stress exponents are all in the range of 2.8 to 4.6 as expected for dislocation-dominated, power-law creep.[24]

Plot of secondary minimum compressive creep rate vs applied stress for (a) 43 pct and (b) 51 pct porous E-Brite tested at 1073 K (800 °C) under flowing Ar. Values in parentheses indicate duration of preoxidation at 1073 K (800 °C) prior to creep testing. For comparison, non-porous E-Brite has a stress exponent of n ≈ 4.0[3]
Significant creep strengthening is observed in the case of pre-oxidized samples, as shown in Figure 6 where the creep rate decreases by about two orders of magnitude after 10 hours of pre-oxidation at 1073 K (800 °C). This behavior is in accordance with the reduction in porosity and growth of the oxide skeleton that strengthen the E-Brite foams. The effect is similar to syntactic foams where additions such as hollow ceramic spheres provide additional creep resistance as the load is transferred from the metallic matrix to the continuous ceramic reinforcement.[25–27] While the contribution of the two effects—reduction in density and growth of oxide—is not specifically detailed in this work, it is apparent that the net effect is well-described by power-law behavior.
Compared to previous creep results on porous, non-pre-oxidized E-Brite at 1123 K (850 °C) in flowing argon,[3] the foams display a somewhat similar stress dependence. In particular, the 42 and 49 pct porous foams without pre-oxidation from previous work exhibited stress exponents of 5.2 and 4.0, respectively. Non-pre-oxidized samples in the present study with 43 and 51 pct porosity have stress exponents of 2.8 and 4.3, respectively (Figure 6). Due to the lower testing temperature used here, these foams also have lower strain rates for a given stress, when compared to their counterpart with 43 and 49 pct porosity.[3]
Electrical Properties
The cross-section of a sample prior to ASR testing is shown in Figure 7 demonstrating that the Ag slurry infiltrates only minimally into the outer layer of sample porosity. Nominal ASR is plotted as a function of temperature in Figure 8 for both heating and cooling, where the area is the nominal sample area, not taking into account porosity. Apparent activation energies, E a = 69 to 82 kJ mol−1 (0.72 to 0.85 eV), were obtained by fitting the data to a modified Arrhenius expression:
where C A is a constant and k is the Boltzmann constant.

SEM micrograph of cross-section of 51 pct porous E-Brite after 24 h oxidation in air at 1123 K (850 °C) and coated with Ag electrode. As viewed here prior to ASR testing, the Ag paint is relatively flat at the surface and does not infiltrate most large pores

Modified Arrhenius plot of ASR vs temperature for dense and porous E-Brite after oxidation at 1123 K (850 °C) for 24 h. Results from a study by Huang et al.[28] on dense E-Brite, pre-oxidized at 1173 K (900 °C) for 24 h, are shown for comparison
As expected, the nominal ASR of the porous samples is lower than that of the fully dense counterpart (Figure 8): since porosity is not taken into account when computing the sample area used for nominal ASR, the reduced surface in contact with Ag leads to an apparently lower nominal ASR for increasingly porous samples. This is evident in Figure 8 where the 51 pct porous foam has a lower nominal ASR as compared to the 43 pct porous sample. Nonetheless, activation energies for samples of both porosities and for the fully dense sample remain in the narrow range of 69 to 82 kJ mol−1 which is similar to the previously reported value of 62 kJ mol−1 for E-Brite obtained upon heating with a similarly applied Ag electrode,[28] These results, therefore, indicate there is no effect from the powder metallurgical processing method or the presence of oxide when determining ASR for interconnects of this type.
While it has been shown that Ag is susceptible to electromigration in the temperature range 943 K to 1153 K (670 °C to 880 °C) under the presence of a large (104 A cm−2) current density,[29] the effects of the small direct current applied (10−2 A cm−2) and short time scale in the present study are not anticipated to affect results. This is confirmed by EDX analysis: a trace at the interface of the electrode and oxidized E-Brite surface shown in Figure 9 after completion of ASR testing indicates that no significant Ag has diffused from the coating. Also, though the applied current may affect oxide growth kinetics, or overlay with possible e.m.f. due to slight differences on oxygen potential or temperature gradients, these effects were not explicitly examined in this work.

SEM micrograph of cross-section and corresponding EDX trace of Ag for a porous E-Brite sample after oxidation for 24 h in air at 1123 K (850 °C), coating with Ag and ASR testing. The abrupt drop in silver at the interface suggests a negligible effect of electromigration for the present conditions
If the oxide scale is dense enough and is able to resist spallation, the continuous Cr2O3 network may decrease the coefficient of thermal expansion of the interconnect, reducing mismatch with the ceramic components. This is an interesting area of further research, also connected to the nascent field of interconnect coatings,[30] which can also mitigate the deleterious effects of oxidation—including chromium evaporation and poisoning—in porous metals for SOFCs.
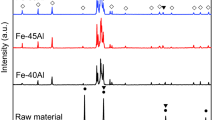
X-ray Diffraction
XRD spectra for as-received E-Brite sheet and the 43 pct porous Fe-26Cr-1Mo sample after 24 hours in air at 1123 K (850 °C) are shown in Figure 10. Results of their non-oxidized counterparts are also shown for comparison. Due to the isostructural nature of Fe2O3 and Cr2O3, it was not possible to distinguish between them. Nonetheless, the oxidation characteristics of the as-received and sintered foam indicate many of the same species are present. This suggests that the commercial E-Brite sheet and the Fe-26Cr-1Mo powder metallurgy alloys, despite their differing processing techniques, exhibit similar oxidation behavior. Some unidentified peaks in the case of Fe-26Cr-1Mo may be due to impurities present in the elemental powders used. One possibility is that the unidentified peaks could potentially be sigma-like precipitates, an intermetallic that develops near grain boundaries that may, in part, contribute to the creep behavior.[10] However, it appears the dominant effect on creep is from oxide formation.

XRD spectra of as-received E-Brite sheet and 43 pct porous Fe-26Cr-1Mo specimens after 0 and 24 h of oxidation at 1123 K (850 °C)
Conclusions
The oxidation kinetics at 1073 K (800 °C) of E-Brite (Fe-26Cr-1Mo, wt pct) foams, fabricated by a salt replication powder metallurgy process and with nearly fully open porosities of 43 and 51 pct, are similar to those of the dense alloy. Increasing porosities leads to higher mass gains due to an increase in surface area; however, parabolic rate constants are unchanged, indicating that the basic mechanism of oxide scale growth is unchanged. The porous samples also have the same narrow range of activation energies for ASR as the dense alloy further confirming that oxidation behavior is not significantly affected by the range of porosities examined in this work.
Upon oxidation at 1073 K (800 °C), mechanical properties of E-Brite foams are significantly influenced by the presence of oxide scale. After just 10 hours of exposure in static laboratory air, the compressive strain rate, at a given stress at 1073 K (800 °C) in argon is reduced by nearly two orders of magnitude, because of the load-bearing capability of the continuous oxide coating the open porosity. This dramatic creep strengthening effect accrues primarily during the initial hours of oxidation, as further oxidation to 100 hours has a smaller strengthening effect: pre-oxidation may thus help ensure more consistent performance of the fuel cell stack throughout its lifetime. This strengthening effect is accompanied by a reduction of open porosity by the choking of fenestrations between pores. Both strengthening and pore filling effects should be taken into account in the design of the stack.
References
Y.B. Matus, L.C. De Jonghe, C.P. Jacobson, S.J. Visco, Solid State Ionics 176, 443–449 (2005)
S. Molin, B. Kusz, M. Gazda, P. Jasinski, J. Power Sources 181, 31–37 (2008)
J.A. Scott, D.C. Dunand, Acta Mater. 58, 6125–6133 (2010)
J.D. Carter, T.A. Cruse, J-M. Bae, J.M. Ralph, D.J. Myers, R. Kumar, and M. Krumpelt: Mater. Res. Symp. Proc., 2003, vol. 756, pp. 545–50
N.P. Brandon, S. Skinner, B.C.H. Steele, Ann. Rev. Mater. Res. 33, 183–213 (2003)
M.C. Tucker, J. Power Sources 195, 4570–4582 (2010)
I. Villarreal, C. Jacobson, A. Leming, Y. Matus, S. Visco, L. De Jonghe, Electrochem. Solid St. 6, A178–A179 (2003)
J. Wu, X. Liu, J. Mater. Sci. Technol. 26, 293–305 (2010)
J.A. Scott, D.C. Dunand, Mater. Sci. Eng., A 585, 32–38 (2013)
Z. Yang, K.S. Weil, D.M. Paxton, J.W. Stevenson, J. Electrochem. Soc. 150, A1188–A1201 (2003)
J.W. Fergus, Mater. Sci. Eng., A 397, 271–283 (2005)
W.J. Quadakkers, J. Piron-Abellan, V. Shemet, L. Singheiser, Mater. High Temp. 20, 115–127 (2003)
P. Bance, N.P. Brandon, B. Girvan, P. Holbeche, S. O’Dea, B.C.H. Steele, J. Power Sources 131, 86–90 (2004)
N.P. Brandon, A. Blake, D. Corcoran, D. Cumming, A. Duckett, K. El-Koury, D. Haigh, C. Kidd, R. Leah, G. Lewis, C. Matthews, N. Maynard, N. Oishi, T. McColm, R. Trezona, A. Selcuk, M. Schmidt, L. Verdugo, J. Fuel Cell Sci. Technol. 1, 61–65 (2004)
N. Oishi and Y. Yoo: in ECS Transactions, Vienna, Austria, 2009, pp. 739–44
P. Blennow, J. Hjelm, T. Klemenso, Å. Persson, K. Brodersen, A. Srivastava, H. Frandsen, M. Lundberg, S. Ramousse, and M. Mogensen: ECS Transactions, Vienna, Austria, 2009, pp. 701–10
M.C. Tucker, G.Y. Lau, C.P. Jacobson, L.C. DeJonghe, S.J. Visco, J. Power Sources 171, 477–482 (2007)
S. Sakuno, S. Takahashi, and H. Sasatsu: ECS Transactions, Vienna, Austria, 2009, pp. 731–37
I. Antepara, I. Villarreal, L.M. Rodriguez-Martinez, N. Lecanda, U. Castro, A. Laresgoiti, J. Power Sources 151, 103–107 (2005)
S. Molin, M. Gazda, B. Kusz, P. Jasinski, J. Eur. Ceram. Soc. 29, 757–762 (2009)
A. Bautista, C. Moral, F. Velasco, C. Simal, S. Guzman, J. Mater. Process. Tech. 189, 344–351 (2007)
Y. Liu, J. Power Sources 179, 286–291 (2008)
W.N. Liu, X. Sun, E. Stephens, M.A. Khaleel, J. Power Sources 189, 1044–1050 (2009)
H.J. Frost and M.F. Ashby: Deformation-Mechanism Maps: The Plasticity and Creep of Metals and Ceramics, Pergamon Press, Oxford [Oxfordshire]; New York, 1982
D.K. Balch, D.C. Dunand, Acta Mater. 54, 1501–1511 (2006)
O. Couteau, D.C. Dunand, Mater. Sci. Eng., A 488, 573–579 (2008)
S. Soubielle, F. Diologent, L. Salvo, A. Mortensen, Acta Mater. 59, 440–540 (2011)
K. Huang, P.Y. Hou, J.B. Goodenough, Solid State Ionics 129, 237–250 (2000)
P.S. Ho, H.B. Huntington, J. Phys. Chem. Solids 27, 1319–1329 (1966)
N. Shaigan, W. Qu, D.G. Ivey, W. Chen, J. Power Sources 195, 1529–1542 (2010)
Acknowledgments
The authors thank Dr. R. Bhat (GE Global Research Center) for useful discussions, D.M. Bierschenk (NU) for his assistance with ASR sample preparation, and Z.D. Feinberg (NU) for his help with XRD analysis. Part of this research was funded by NASA through a subcontract from GE (Award NNC06CB31C). JAS also thanks Bell Labs, the National Science Foundation, and the Northwestern University Nanoscale Science & Engineering Center for graduate research fellowships.
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript submitted February 15, 2014.
Rights and permissions
About this article

Cite this article
Scott, J.A., Dunand, D.C. Effect of Oxidation on Creep Strength and Resistivity of Porous Fe-26Cr-1Mo. Metallurgical and Materials Transactions E 1, 303–310 (2014). https://doi.org/10.1007/s40553-014-0031-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40553-014-0031-8