
Abstract
Objective
The aim of this clinical trial was to establish the bioequivalence of two tablets containing acetaminophen 650 mg (reference) and acetaminophen 650 mg plus caffeine 65 mg (test), administered orally, in fasting conditions in healthy Mexican volunteers.
Methods
Blood samples were taken from 21 male and five female individuals, during a 24-h period, to characterize the pharmacokinetic profile of acetaminophen. Plasma samples were quantified by ultra-performance liquid chromatography, tandem mass spectrometry. Pharmacokinetic metrics (maximum plasma concentration, area under the curve from time zero to the last sampling time, and area under the curve from time zero to infinity) were used to determine the 90 % confidence interval of the test/reference coefficient.
Results
The geometric mean values for maximum plasma concentration obtained for the reference and test products were 9.46 ± 34.21 and 9.72 ± 32.38 µg/mL, respectively, whereas for the area under the curve from time zero to the last sampling time the values obtained were 34.93 ± 32.58 and 35.89 ± 31.03 µg h/mL for the reference and test formulations, respectively. The 90 % confidence intervals were within the acceptance range (80–125 %).
Conclusions
The test product was bioequivalent to the reference product. A faster absorption was seen in the test formulation in the Mexican population.
Similar content being viewed by others

In this clinical trial, the test product containing acetaminophen 650 mg plus caffeine 65 mg was bioequivalent to the reference product containing only acetaminophen 650 mg, in the Mexican population. |
The absorption rate of acetaminophen was slightly faster in combination with caffeine, only in the first sample schedule; according to Renner et al., this might mean a better analgesic effect than monotherapy in the Mexican population. |
1 Introduction
In Mexico, there are currently more than 50 products containing acetaminophen, either alone or in combination with other drugs, in different doses and pharmaceutical forms. However, no pharmacokinetic studies regarding these products in the Mexican population are available in the scientific literature. Therefore, it is important to perform clinical trials to increase the knowledge on the bioavailability of this drug and its combinations in Mexican individuals.
Acetaminophen is an analgesic, antipyretic, and anti-inflammatory drug that inhibits cyclooxygenase, preventing the synthesis of prostaglandins [1–5]. Its mechanism of action is similar to aspirin; however, it does not affect platelet aggregation, exert cardiovascular effects, cause respiratory actions, or result in gastric damage [1–6]. The oral absorption of acetaminophen is fast and complete [5]; meals reduce its final absorption, the volume of distribution is 0.95 ± 0.12 L/kg, it crosses the placental barrier, and it can appear in human milk. Acetaminophen has a low level of plasma protein binding (10–30 %) [1]. Its hepatic metabolism is extensive (95 %) and presents a first-pass effect [1, 4, 5, 7]. The clearance of acetaminophen is 5 mL/min/kg, the renal excretion as unchanged drug is only 1–4 %, and the rest is eliminated as inactive metabolites. Its elimination half-life is between 2 and 3 h [1].
Acetaminophen is used for the temporary relief of minor aches and pains associated with colds, headache, toothache, muscular aches, and backache. Additionally, it is used for minor arthritis pain, menstrual cramps, and for fever reduction [8]. Acetaminophen 1000 mg is an effective and well-tolerated treatment for episodic and moderate migraine headache. In addition, it provides a beneficial effect on associated symptoms (nausea, photophobia, phonophobia, and functional disability) [9]. Acetaminophen alone is not an effective therapy for acute migraine but is the first choice during pregnancy. The combination of an analgesic containing aspirin, caffeine, and acetaminophen is an effective first-line treatment for migraine [10].
Caffeine is an effective adjuvant analgesic [2] and potentiates the analgesic effect of acetaminophen (which alone requires a 40 % higher dose to reach a level of pain relief compared with that obtained with caffeine). Caffeine acts as a mild psycho-stimulant, being able to re-establish the alertness and performance capacity in subjects experiencing fatigue [1, 11]. The hepatic metabolism produces active metabolites (paraxanthine, theophylline, and theobromine) that contribute to pharmacologic effects [1, 11–13]. Caffeine contributes to competitive inhibition of phosphodiesterase by inactivating the cyclic adenosine monophosphate. It also increases the permeability of calcium in the sarcoplasmatic reticulum and blocks the adenosine receptors, thereby increasing the activity of neurotransmitters such as acetylcholine, adrenaline, noradrenaline, dopamine, and glutamate [1]. The oral absorption of caffeine is complete and fast, needing 1 h to reach the maximum plasma concentration (t max). After an oral dose of 1 mg/kg, its bioavailability is 99 % and the peak serum concentration is 1–2 µg/mL [1]. Its volume of distribution is 0.55 L/kg, reaching all tissues; thus, it crosses the blood–brain and placental barriers, is found in mother’s milk, and only 35 % was bound to plasma proteins [1]. Caffeine has a linear elimination, with an elimination half-life of approximately 5 h [1].
2 Methods
2.1 Subjects
The study was approved by the Research and Ethic Committees and the Federal Commission for the Sanitary Risk Protection of Mexico (Comisión Federal para la Protección de Riesgos Sanitarios, Mexico). It also followed the Good Clinical Practices Guidelines [14], the ethical principles for medical research for humans stated in the Declaration of Helsinki [15], the General Health Law of Mexico (Ley General de Salud, México) [16], and the Regulations of the General Health Law for Health Research (Reglamento de la ley General de Salud en Materia de Investigación para la Salud, Mexico) [17]. Additionally, this study was conducted according to the Mexican Official Norm (NOM-177-SSA1-2013) [18].
Twenty-six Mexican subjects were included in this study and were determined as healthy by clinical and laboratory tests. All of them were informed regarding the possible risks and adverse events after taking the study product. All the volunteers gave their informed written consent to participate in the study.
2.2 Study Design
The study was conducted according to an open, randomized, single-dose crossover design, performed under fasting conditions. Two periods, two sequences, with a wash-out period of 7 days were used to analyze the pharmacokinetic metrics of the two products. The sample size was determined considering an expected power of 80 % and the intra-subject variability of acetaminophen reported in the literature [19]. The test product was acetaminophen plus caffeine tablets of 650 mg plus 65 mg (Sedalmerck Max®; Merck, S.A. de C.V., Mexico [M31344]), whereas the reference product was acetaminophen tablets of 650 mg (Tempra® Forte; Bristol-Myers Squibb de México, Mexico, S. de R.L. de C.V., Mexico [3G100331]), one tablet administered to the volunteers in two different periods. The sample schedule was 0.00, 0.25, 0.50, 0.75, 1.00, 1.50, 2.00, 2.50, 4.00, 6.00, 8.00, 10.00, 12.00, and 24.00 h after the dose. The blood samples were obtained with heparin tubes, the plasma was separated by centrifugation (2500g × 5 min at 4 ± 2 °C) and stored at a temperature below −40 °C until the analysis. The safety of the subjects was monitored during the whole study.
2.3 Analytical Quantification
The analytical procedure for the quantification of acetaminophen in plasma was developed and validated within the parameters of selectivity, precision, accuracy, linearity, and sensitivity. One hundred microliters of human plasma was added to 50 µL of internal standard (zidovudine 20 µg/mL; US Pharmacopeia reference standard, lot number HOF263). Then, 0.1 mL of formic acid (100 mM; J.T. Baker, lot number 0000071580) and 1.0 mL of ether anhydrous were added to all samples to perform a liquid-liquid extraction (J.T. Baker; lot number 0000075778) that was agitated for 2 min at 5000 rpm (Precellys® 24, Bertin Corp, Rockville MD, USA). The samples were centrifuged for 5 min at 14,000 rpm at 0 °C and stored at a temperature below −70 °C. The organic phase was transferred into glass tubes (13 × 100 mm), placed in a water bath at 30 °C, and evaporated under a nitrogen current, until dry. The samples were reconstituted with 400 µL of an acetonitrile:formic acid solution (10 mM) 90:10 v/v and centrifuged at 2500 rpm for 1 min. The samples were transferred into vials for an autosampler and 5 µL was injected into the ultra-performance liquid chromatography (UPLC)-tandem mass spectrometer using a Xevo TQ-D (Waters Corp, Milford MA, USA) equipped with electrospray ionization. The UPLC was an Acquity UPLC-H Class (Waters Corp, Milford MA, USA), with a quaternary pump, an autosampler with controlled temperature, a column oven, and an in-line degasifier. The data were processed using the MassLynx 4.1 software (Waters Corp, Milford MA, USA).
The chromatographic separation was performed by a Cortecs C18 column (4.6 × 100 mm, 2.7 µm; Waters, Spain) and the mobile phase consisted of an acetonitrile:formic acid solution (10 mM) 85:15 v/v at a flow rate of 0.7 mL/min. The autosampler was kept at temperature of 6 °C, the injection volume was 5 µL, the run time for each injection was 1.5 min, and the retention times were 1.18 min for acetaminophen and 1.19 min for zidovudine. The detection was performed in positive mode by the monitoring of multiple reactions in the mass spectrometer Xevo TQ-D (Waters Corp, Milford MA, USA) Waters triple quadrupole equipped with electrospray ionization. The monitored transitions were m/z 152.0 → 110.0 for acetaminophen and m/z 268.0 → 127.0 for zidovudine. The temperature of the source was 550 °C and the voltage of the capillary was 2.4 kV. The voltage of the cone for acetaminophen was 35 V and for zidovudine was 14 V. The energy of optimal collision for acetaminophen was 15 V and for zidovudine was 8 V, argon was the collision gas employed.
2.4 Pharmacokinetics and Statistical Analysis
The pharmacokinetics of both products was determined by a non-compartmental analysis, using the Phoenix® WinNonlin® 6.3 Centara L.P. software, USA. The C max and t max were determined by analyzing the concentration profiles vs. time. The determination of area under the curve from time zero to the last sampling time (AUC0–t ), was performed by the trapezoidal rule. The constant of elimination (K el) was determined from the linear terminal part of the data logarithmically transformed and was estimated through a simple linear regression analysis considering at last three different concentrations from the t max value. It was used to determine the area under the curve from time zero to infinity (AUC0–∞) according to the equation: AUC0–∞ = AUC0–t + C t /K el, where C t is the concentration at the last time of sampling used. The elimination half-life was determined by the ratio of In(2)/K el. Partial AUCs were calculated at 0.5, 1.0, 1.5, 2.0, 3.0, and 4.0 h, by linear trapezoidal interpolation, which is applied to each pair of consecutive points in the data set. Only the 3.0-h time has an endpoint that was not included in the data set, thus the trapezoidal linear interpolation rule was used to add a concentration value for that endpoint.
The bioequivalence between both products was determined by calculation of the confidence intervals (CIs) at 90 % of the logarithm of the coefficient test/reference such as C max, AUC0–t , and AUC0–∞ in terms of acetaminophen pharmacokinetic metrics.
Statistical analysis of the partial AUCs was by calculation of the CIs at 90 % of the logarithm of the coefficient test/reference. Analysis of variance was determined to evaluate the effects on the formulation variation, sequence, and period. Both were calculated with Phoenix® WinNonlin® 6.3.
3 Results
3.1 Study Population
Twenty-six Mexican volunteers (five female and 21 male individuals) were included in this study, with ages between 18 and 45 years (23.9 ± 5.1 years) and a body weight between 44.2 and 82.1 kg (60.3 ± 9.3 kg). The body mass index of participants was 22.7 ± 2.3 kg/m2.
3.2 Validation of the Analytical Method
The analytical method was linear in the range of 0.1–30 µg/mL, the best linear fit and least-squares residuals for the calibration curve were achieved with a 1/x 2 weighing factor, giving a mean linear regression equation for the calibration curve of: y = 1.6 x + 5.9 × 10−3, where y is the peak area ratio of acetaminophen to zidovudine and x is the concentration of the analyte. The mean correlation coefficient of the weighted calibration curve generated during the validation was 0.99.
Table 1 shows the results of the evaluation of precision and accuracy of the analytical method, meeting the acceptance criteria of ±15 and ±20 % of the lower limit of quantification. There were no significant matrix effects for acetaminophen or zidovudine. The stability of acetaminophen in human plasma under different temperatures was validated under the typical plasma storage and processing conditions used throughout the current study such as the autosampler temperature, room temperature, refrigeration temperature, freeze-thaw cycles, and at least 63 days of long-term storage conditions (−70 °C).
3.3 Pharmacokinetic Analysis
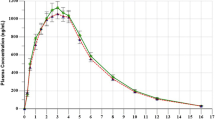
Figure 1 shows the mean plasma concentrations vs. time of the arithmetic data transformed semi-logarithmically.

Geometric mean plasma concentrations vs. time in linear scale (a) and semi-logarithmic scale (b), following the administration of the test and reference products. Error bars present ±1 geometric standard deviation
Results of the pharmacokinetic analysis after oral administration of 650 mg of acetaminophen (reference product) and 650 mg of acetaminophen plus 65 mg of caffeine (test product) in 25 subjects that completed the study are summarized in Table 2.
In Table 3, the results of the 90 % CI of the ratio of the geometric means considering the intra-subject variability are shown. As observed, the values of the CI are within the range of acceptance (80–125 %).
Table 4 shows the results of the equivalence of partial AUCs at different time points between the two formulations. The t max values are shown in the boxplot in Fig. 2, evaluated by the Wilcoxon Mann–Whitney test with a significance level of α = 0.05 and a p value of 0.49. It shows one outlier is present in the reference product; however, it only represents an extreme value without statistical significance.

Pharmacokinetic metric t max (A reference product, B test product). t max time to maximum plasma concentration
4 Discussion
The final number of evaluated subjects was 25, one volunteer was excluded because of receiving concomitant medication (hydrocortisone, loratadine, and chlorpheniramine) for the treatment of a non-serious adverse event not related to the study (food allergy). Another volunteer had a flu condition, with a total of two non-serious adverse events during the study (the last volunteer finished the trial).
The method showed good precision and accuracy. Table 1 summarizes the intra- and inter-day precision and accuracy values for acetaminophen from quality-control samples. In this assay, the intra- and inter-assay precisions were measured to be below 5.0 and 11.5 %, respectively, with relative errors from −6.3 to 7.3 %. These values were within the acceptable range, and the method was thus judged to be suitably accurate and precise. Under the present low limit of quantification of 0.1 ng/mL, this is sensitive enough to investigate the pharmacokinetic behavior of acetaminophen in clinical studies.
The pharmacokinetic analysis showed no differences in the absorption rate and bioavailability of the drug to reach the bloodstream. Table 2 shows the geometric mean C max values of 9.46 and 9.72 µg/mL for the reference and test products, respectively. The geometric mean AUC0–t values were 35.89 h µg/mL and AUC0–∞ 37.29 h µg/mL for the reference product, while for the test product the values were 34.93 and 36.22 h µg/mL, respectively. These values demonstrated that the bioavailability of the drug in both formulations did not present differences, which was confirmed with the results in Table 3. Indeed, at a 90 % CI (range 80–125 %), the bioequivalence was demonstrated. The intra-subject variability obtained was of 22.0 % for C max, 8.2 % for AUC0–t , and 7.3 % for AUC0–∞ (Table 3), demonstrating that acetaminophen in both formulations presents a low variability in the Mexican population.
Table 4 shows the statistical analysis of the partial AUCs determined at different times. The partial AUCs from 0 to 2.0 h show differences because they are not contained in the 90 % CI range (80–125 %) and there is high variability in the results. This denotes higher values for the test product than the reference product. This behavior is related to differences in absorption that were present only during the period from time zero until 120 min post-dose. They can be attributed to the presence of caffeine, according to the investigation performed by Renner et al. [19], where it was demonstrated that caffeine presence assures the rapid absorption of acetaminophen. Moreover, another study, which compared the administration of conventional acetaminophen tablets with effervescent tablets, showed that there is also a modification in t max, suggesting that changes in the conditions of the media in which acetaminophen is dissolved favor its rapid absorption [20]. Assuming that the products used in this clinical trial had no differences between formulations (both are immediate-release tablets, without film, enteric, or sugar coatings), the difference is that the test product contains acetaminophen plus caffeine while the reference product only contains acetaminophen.
The t max was evaluated by the Wilcoxon Mann–Whitney test, obtaining a p value of 0.49 (p > 0.05), demonstrating there are no statistical differences in the t max value between both formulations. For this reason, the C max did not suffer modification because t max is not an adequate indicator of the absorption rate [20]. It was expected to observe differences in the pharmacokinetics with the presence of caffeine, as indicated by Renner et al. [19]. Indeed, in this study, the 90 % CI of partial AUCs allowed us to evaluate the difference in the plasma concentrations of acetaminophen, showing that the bioavailability and absorption rate of acetaminophen with the test product was greater than the reference product, only in the first minutes. This determination was based on the US Food and Drug Administration guidelines, “Bioavailability and Bioequivalence Studies for Orally Administered Drug Products: General Considerations” [21], which recommend the determination of “early exposure” to evaluate the information regarding the appropriate clinical efficacy/safety in pharmacokinetic/pharmacodynamic studies.
5 Conclusions
With the results obtained it was concluded that the test product is bioequivalent to the reference formulation in terms of rate and bioavailability of acetaminophen because the 90 % CIs are within the acceptance range of 80–125 %. It was also concluded that the determination of the 90 % CI of partial AUCs is more sensitive to establish the degree of absorption of a drug in the first hours following its oral administration than the statistical analysis of t max. However, it has be used with more than three sampling points before t max and requires extreme sample sizes owing to its high variability. In the Mexican population studied, the test product is absorbed slightly faster than the reference product, thus an analgesic effect can be expected in a shorter period of time, which represents an advantage in the management of acute pain.
References
Brunton LL, Lazo JS, Parker KL (eds.). Goodman and Gilman’s the pharmacological basis of therapeutics, 12th edn. New York: McGraw-Hill; 2006. (Section IV, Chapter 34, p. 982–4 and Table AII-1 Pharmacokinetic Data, p. 1899).
Liu DJ, Kotler M, Sharples S. Pharmacokinetic and bioequivalence study evaluating a new paracetamol/caffeine formulation in healthy human volunteers. J Bioequiv Availab. 2011;3:11.
Anderson BJ, Holford N. Pharmacokinetics and pharmacodynamics of analgesic drugs. Pain in neonates and infants. Ch09-N52061.qxd 2/7/07. Elsevier, Edinburgh Scotland.
Crawford V. Pharmacodynamic/pharmacokinetic of aspirin and acetaminophen: comparison between aspirin (asa) and acetaminophen (apap). Drugs Toxicol. 2014.
DrugBank. Acetaminophen. DB00316 (APRD00252). https://www.drugbank.ca/drugs/DB00316. Accessed 29 Sep 2106.
Ortega-Barrales P, Padilla-Weigand R, Molina-Díaz A. Simultaneous determination of paracetamol and caffeine by flow injection-solid phase spectrometry using C18 silica gel as a sensing support. Anal Sci. 2002;18(11):1241–6.
Shinoda S, Aoyama T, Aoyama Y, et al. Pharmacokinetics/pharmacodynamics of acetaminophen analgesia in Japanese patients with chronic pain. Biol Pharm. 2007;30(1):157–61.
Prior MJ, Codispoti JR, Fu M. A randomized, placebo-controlled trial of acetaminophen for treatment of migraine headache. Headache. 2010;50(5):819–33.
PubMed Health. Medication for migraines. http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0072555/. Accessed 29 Sep 2016.
Gilmore B, Michael M. Treatment of acute migraine headache. Am Fam Physician. 2011;83:271–80.
Syeda SA, Kamimorib GH, Kelly WB, Eddington ND. Multiple dose pharmacokinetics of caffeine administered in chewing gum to normal healthy volunteers. Biopharm Drug Dispos. 2005;26:403–9.
Lelo A, Birkett DJ, Robson RA, Miners JO. Comparative pharmacokinetics of caffeine and its primary demethylated metabolites paraxanthine, theobromine and theophylline in man. Br J Clin Pharmacol. 1986;22:177–82.
DrugBank. Caffeine. DB00201 (APRD00673). http://www.drugbank.ca/drugs/DB00201. Accessed 29 Sep 2016.
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH harmonised tripartite guideline: guideline for good clinical practice E6(R1), June 1996.
Ethical principles for medical research in humans of the Declaration of Helsinki issued by the 64th General Assembly, Fortaleza, Brazil, October 2013.
General Health Law of Mexico (Ley General de Salud, México).
Regulations of the General Health Law for Health Research (Reglamento de la Ley General de Salud en Materia de Investigación para la Salud, México).
Norma Oficial Mexicana NOM-177-SSA1-2013. Que establece las pruebas y procedimientos para demostrar que un medicamento es intercambiable. Requisitos a que deben sujetarse los Terceros Autorizados que realicen las pruebas de intercambiabilidad. Requisitos para realizar los estudios de biocomparabilidad. Requisitos a que deben sujetarse los Terceros Autorizados, Centros de Investigación o Instituciones Hospitalarias que realicen las pruebas de biocomparabilidad.
Renner B, Clarke G, Grattan T, et al. Caffeine accelerates absorption and enhances the analgesic effect of acetaminophen. J Clin Pharmacol. 2007;47:715–26.
Rygnestad T, Zahlsen K, Samdal FA. Absorption of effervescent paracetamol tablets relative to ordinary paracetamol tablets in healthy volunteers. Eur J Clin Pharmacol. 2000;56:141–3.
FDA. Bioavailability and bioequivalence studies for orally administered drug products: general considerations. 2000. http://www.fda.gov/ohrms/dockets/ac/03/briefing/3995B1_07_GFI-BioAvail-BioEquiv.pdf. Accessed 9 Feb 2016.
Acknowledgments
The authors and the trial staff both contributed to the manuscript. Assistance with the writing of the submission was provided by Nora Angélica Núñez Guzmán and the staff of Clinica de Enfermedades Crónicas y de Procedimientos Especiales (CECYPE). All named authors meet the International Committee of Medical Journal Editors criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval of the version to be published.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was performed entirely by CECYPE and was supported by Merck, S.A. de C.V. Mexico.
Conflict of interest
Nora Angélica Núñez Guzmán, Daniel Ruiz Molina, Benigno Figueroa Núñez, Juan Carlos Soto-Sosa, and Jorge Eduardo Herrera Abarca are employed by CECYPE and declare no conflicts of interest.
Ethics approval and consent to participate
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Declaration of Helsinki. Informed consent was obtained from all subjects included in the clinical trial.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Guzmán, N.A.N., Molina, D.R., Núñez, B.F. et al. Bioequivalence and Pharmacokinetic Evaluation Study of Acetaminophen vs. Acetaminophen Plus Caffeine Tablets in Healthy Mexican Volunteers. Drugs R D 16, 339–345 (2016). https://doi.org/10.1007/s40268-016-0145-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-016-0145-9