
Abstract
Daratumumab is a CD38 monoclonal antibody recently approved for the treatment of multiple myeloma (MM). We report daratumumab pharmacokinetic data from GEN501, a phase I/II dose-escalation (0.005–24 mg/kg) and dose-expansion (8 or 16 mg/kg) study, and SIRIUS, a phase II study (8 or 16 mg/kg), in relapsed or refractory MM. Noncompartmental analysis was conducted to characterize daratumumab pharmacokinetics, and, in both studies, daratumumab exhibited nonlinear pharmacokinetic characteristics. Decreasing daratumumab clearance with increasing dose suggests saturation of target-mediated clearance at higher dose levels, whereas decreasing clearance over time with repeated dosing may be due to tumor burden reductions as CD38-positive cells are eliminated. These and other pharmacokinetic data analyses support the use of the recommended dose regimen of daratumumab (16 mg/kg weekly for 8 weeks, every 2 weeks for 16 weeks, and every 4 weeks thereafter) to rapidly saturate target-mediated clearance during weekly dosing and maintain saturation when dosing every 2 or 4 weeks.
Similar content being viewed by others


Daratumumab is a CD38 monoclonal antibody that was recently approved in the US and Canada for the treatment of patients with multiple myeloma (MM) who received three or more prior treatments, including a proteasome inhibitor (PI) and an immunomodulatory drug (IMiD), or who are double-refractory to these agents. In Europe, the drug is approved for relapsed and refractory MM patients whose prior therapy included a PI and an IMiD, and who demonstrated disease progression on the last therapy. |
Following intravenous infusion, daratumumab exhibits nonlinear pharmacokinetic characteristics, consistent with target-mediated drug disposition. |
Administration of daratumumab 16 mg/kg weekly for 8 weeks rapidly saturates target-mediated clearance; dosing every 2 weeks for 16 weeks, and every 4 weeks thereafter is sufficient to maintain target saturation. |
1 Introduction
Outcomes in patients with multiple myeloma (MM) have improved significantly in the past two decades [1–3], largely due to the availability of novel immunomodulatory drugs (IMiDs) such as thalidomide, lenalidomide, and pomalidomide, as well as proteasome inhibitors (PIs) such as bortezomib [1–3]. Indeed, these treatments are now considered the standard of care for patients with newly diagnosed MM, either as monotherapy or in combination with traditional therapies or with one another [4, 5]. However, treatment options for patients with MM whose disease has relapsed or is refractory to IMiDs and/or PIs are limited, and prognosis is poor [6–8]. Therefore, novel therapies are urgently needed for this difficult-to-treat population.
Daratumumab is a first-in-class, anti-CD38 human immunoglobulin (Ig) G1 monoclonal antibody that was recently approved as monotherapy for patients with MM who received three or more prior treatments, including a PI and an IMiD, or who are double-refractory to a PI and an IMiD [9]. CD38 is highly and uniformly expressed on myeloma cells [10, 11], making it a relevant target for the treatment of MM. The anti-myeloma activity of daratumumab is mediated via several mechanisms of action, including complement-dependent cytotoxicity, antibody-dependent cell-mediated toxicity, antibody-dependent cellular phagocytosis, and apoptosis via crosslinking and modulating ectoenzyme [12, 13]. Subsets of CD38-expressing regulatory T and B cells, as well as myeloid-derived suppressor cells, undergo lysis following treatment with daratumumab [14]. As a result of these changes in immune cell function, increased CD4+ and CD8+ T-cell absolute counts and total lymphocyte percentages have been observed in both bone marrow and peripheral blood [14].
The clinical efficacy and safety of daratumumab monotherapy have been demonstrated in two open-label studies in patients with relapsed or refractory MM. In the phase I/II GEN501 study (ClinicalTrials.gov identifier NCT00574288), 36% of patients receiving the recommended dose of daratumumab 16 mg/kg had a partial response or better and the 1-year overall survival rate was 77% [15]. In the phase II SIRIUS study (NCT01985126), the overall response rate (ORR) was 29% and the 1-year overall survival rate was 65% in a population of heavily pretreated MM patients, most of whom were double-refractory to bortezomib and lenalidomide [16]. In both studies, daratumumab demonstrated a favorable safety profile; the most frequently reported infusion-related reactions were predominantly respiratory issues (e.g. nasal congestion, cough, throat irritation), were grade 1 or 2 in severity, occurred during the first infusion, and were manageable [15, 16].
Dose optimization is a challenge for novel therapies. As the first mAb approved for MM, understanding the pharmacokinetic profile of daratumumab is critical to determining an efficacious and safe dose and dosing schedule. The aim of this study was to characterize the pharmacokinetics of daratumumab, administered via intravenous infusion, using data collected from the GEN501 and SIRIUS studies in patients with MM relapsed from or refractory to prior PIs and/or IMiDs.
2 Methods
Data were collected from two separate clinical studies—GEN501 (NCT00574288) and SIRIUS (NCT01985126). The individual study protocols were reviewed by an independent Ethics Committee or Institutional Review Board at each study site, and the studies were conducted according to the principles of the Declaration of Helsinki and the International Conference on Harmonisation Good Clinical Practice guidelines. All participants provided written informed consent prior to any study-specific procedures.
2.1 Patients and Study Designs
2.1.1 Gen501
GEN501 was a phase I/II, open-label, multicenter study comprising two parts (Fig. 2a); Part 1 was a dose-escalation study, and Part 2 was a single-arm dose-expansion study involving multiple patient cohorts, with dosing of daratumumab based on the results of Part 1. A detailed description of this study has been published elsewhere [15]. Briefly, eligible patients were ≥18 years of age, had an Eastern Cooperative Oncology Group performance status score of ≤2, had documented myeloma requiring systemic therapy, and had relapsed from or were refractory to two or more prior therapies, including PIs, IMiDs, chemotherapy, and autologous stem cell transplantation.
In Part 1, sequential cohorts received intravenous doses of daratumumab ranging from 0.005 to 24 mg/kg, administered over 6–8 h.
Based on the findings from Part 1, five cohorts were enrolled in Part 2 (Fig. 2a). In three of the cohorts, daratumumab was administered in Part 2 at 8 mg/kg weekly for 8 weeks, every 2 weeks for 16 weeks, and every 4 weeks until disease progression.
2.1.2 SIRIUS
SIRIUS, a phase II, open-label, international, multicenter study (Fig. 2b), is described in detail elsewhere [16]. Inclusion criteria were similar to those for the GEN501 studies, with the exception that patients were required to have progressed on their most recent line of therapy and had received three or more prior lines of therapies, including a PI and an IMiD, or were double-refractory to both a PI and an IMiD.
In Part 1, patients were randomized to daratumumab 8 mg/kg every 4 weeks or 16 mg/kg weekly for 8 weeks, followed by every 2 weeks for 16 weeks and every 4 weeks thereafter. Based on the cumulative response data, the 16 mg/kg dose was established as the recommended dose regimen and was administered to the expansion cohort in Part 2.
2.2 Pharmacokinetic Analysis
In Part 1 of GEN501, blood sampling for pharmacokinetic analysis was performed before and after the first pre-dose; before the first full infusion and at the end of infusion (EOI), EOI + 2 h, EOI + 5 h, and EOI + 24 h; any time during days 4, 8, and 15; before the second pre-dose (day 21); on days 22, 29, 36, 43, and 50 before the weekly daratumumab infusion; before the last full infusion and at EOI, EOI + 2 h, EOI + 5 h (optional), and EOI + 24 h; and at any time on days 61 and 65 and weeks 10, 12, 16, 20, 24, and 28 from the start of treatment. In Part 2 of GEN501, sampling time points varied for different cohorts, but generally sampling was performed at screening, before the first full infusion, at EOI, and at EOI + 2 h; before infusion and EOI during weekly visits; and at 1, 2, 4, and 6 weeks after the last infusion.
In the SIRIUS study, pharmacokinetic sampling was performed at pre-dose and EOI time points for the 16 mg/kg dose group, whereas those receiving daratumumab 8 mg/kg (administered once every 4 weeks) had more intense sampling following the first full infusion, allowing estimation of pharmacokinetic parameters.
In GEN501, the median dose intensity was ≥90% of the target dose in Part 1 and 100% in Part 2. In SIRIUS, the median relative dose intensity was 100% of the target dose. However, samples from GEN501 and SIRIUS were excluded from the pharmacokinetic analysis if they were taken following a dose in which <80% of the intended dose was administered.
Venous blood samples (5 mL) were collected, and an enzyme-linked immunosorbent assay (ELISA; lower limit of quantification × minimum required dilution = 200 ng/mL; BioAnalytical Research Corporation, Ghent, Belgium, and, subsequently, Janssen Research & Development, LLC, Spring House, PA, USA) was used to determine serum daratumumab concentrations in both studies. The ELISA method was validated according to criteria established in the Guidance for Industry: Bioanalytical Method Validation [17]. Specifically, a 96-well microplate was coated with CD28 protein overnight at 4°C, blocked with assay buffer for 1 h at room temperature, and washed three times. Standard curve calibrators were prepared in an assay buffer containing 2% human serum. Quality control and study samples were thawed, equilibrated to room temperature, and diluted 1:50 before adding the samples to the microplate. The microplate was then incubated at room temperature for 2 h with continuous shaking and washed three times with assay buffer. HRP-conjugated mouse anti-human IgG1 Fc antibody was added to the microplate, which was then covered with aluminum foil and incubated for 1.5 h at room temperature with shaking. The plate was then washed and incubated with azino benzothiazoline sulphonic acid solution for 30 min. The colorimetric reaction was stopped by adding 2% oxalic acid, and the optical density was read at 405 nm. Daratumumab concentrations were determined by interpolation from the standard curve using a nonlinear logistic curve-fit-based regression model.
2.3 Statistical Analysis
In both studies, serum pharmacokinetic parameters were estimated from the concentration and actual time data using noncompartmental analysis implemented in Phoenix™ WinNonlin® software (Pharsight, St. Louis, MO, USA). For the estimation of pharmacokinetic parameters, concentrations below the limit of quantification were treated as zero if no previous value was quantifiable, or if at least one previous value was measurable and at least one later value was available; otherwise, the value was treated as missing.
For Part 1 of the GEN501 study, only the concentration data immediately before and after the first and seventh full infusions were included in the pharmacokinetic parameter analysis. Descriptive statistics were used to summarize all data, including maximum concentration (C max), half-life (t ½), total systemic clearance of drug after intravenous administration, and volume of distribution (V Z). The area under the concentration–time curve (AUC) was computed by the linear trapezoidal with linear interpolation method, and the terminal elimination rate constant was estimated using the best-fit method (i.e. regression of the natural logarithm of concentration versus sampling time) and uniform weighting. In Part 2 of the GEN501 study, pharmacokinetic parameters (as previously stated) were derived only for the first and last doses of daratumumab.
In the SIRIUS study, no pharmacokinetic parameters were derived for the daratumumab 16 mg/kg group due to sparse sampling; sufficient data were available for the 8 mg/kg cohort to calculate pharmacokinetic parameters after the first dose. AUC values were calculated using a combination of linear and logarithmic trapezoidal methods, and the terminal rate constant associated with the terminal portion of the serum concentration versus time curve was determined by least-squared regression analysis of the log-linear portion of the terminal phase with the adjusted coefficient of determination.
In the GEN501 study, the pharmacokinetic analysis set comprised all treated patients with at least one baseline and one post-baseline pharmacokinetic assessment. In the SIRIUS study, the pharmacokinetic analysis set consisted of all treated patients with at least one post-infusion sample.
3 Results
Pharmacokinetic data were available from 2572 measurable samples from 223 patients across both studies: 100 patients (of 104 enrolled; 32 from Part 1 and 72 from Part 2) in the GEN501 study and 123 patients (of 124 enrolled) in the SIRIUS study (electronic supplementary Table 1). Of the 223 patients who provided samples included in the pharmacokinetic analysis, 150 received daratumumab 16 mg/kg. The baseline demographics and clinical characteristics of the patients included in the pharmacokinetic analysis are presented in electronic supplementary Table 2. The dose and dosing schedule for the GEN501 and SIRIUS studies have been reported in detail elsewhere [15, 16] and are summarized in electronic supplementary Fig. 1a, b, respectively.
3.1 GEN501 Study
In Part 1 of the GEN501 study, 32 patients received the first full infusion of daratumumab and 16 patients received the last (seventh) full infusion. All serum daratumumab concentrations for patients receiving doses of 0.005 or 0.05 mg/kg were below the limit of quantification. Following the first full infusion, the daratumumab C max was approximately dose proportional for doses of 1–24 mg/kg (Figs. 1a, 2a; Table 1). C max increased in a ratio of 1:2:4:8:20:25 as the daratumumab dose increased in a ratio of 1:2:4:8:16:24; however, C max increased in a greater than dose-proportional manner after the last (seventh) dose (Figs. 1a, 2b; Table 1). The AUC from time zero extrapolated to infinity (AUCinf) increased in a greater than dose-proportional manner after both the first and last doses (Fig. 1b). In the 16 mg/kg cohort, mean ± standard deviation (SD) C max was 405.75 ± 72.50 μg/mL after the first full infusion and 993.65 ± 127.04 μg/mL after the last infusion, whereas mean ± SD AUCinf was 56,893.56 ± 22,030.42 and 371,159.32 μg·h/mL after the first and last infusions, respectively. Based on elimination t ½ after the last full infusion, it was unlikely that most patients in Part 1 had reached steady state by the last infusion. Daratumumab elimination showed nonlinear characteristics: t ½ increased with increasing dose and with multiple doses, whereas clearance decreased with increasing dose and with multiple doses, particularly in the groups receiving ≥2 mg/kg of daratumumab (Table 1). Mean ± SD clearance after the first 16 mg/kg dose in Part 1 of the GEN501 study was 0.32 ± 0.13 mL/h/kg (n = 3), whereas, following the seventh dose, clearance decreased to 0.10 mL/h/kg (n = 1). The mean V Z varied (mean 31.90–104.77 mL/kg) across doses and dose levels, but was reasonably consistent among the dose groups for both the first and last full infusions. The mean V Z for the 16 mg/kg group was 45.22 mL/kg after the first full infusion and 31.90 mL/kg after the last full infusion. These V Z values indicate that daratumumab is confined primarily in the vascular system.

a Mean Cmax and b mean AUC across dose cohorts in Part 1 of the GEN501 study. Error bars represent ± standard deviation. AUC area under the concentration–time curve from time zero extrapolated to infinity, C max maximum concentration

Mean daratumumab serum concentration by dose group in Part 1 of the GEN501 study for a the first full infusion and b full infusions 2–7. Error bars represent mean ± 1 standard deviation. A pre-dose of daratumumab (10% of the full dose; maximum 10 mg daratumumab in total) was administered on day 0, prior to the first full infusion, which was administered on day 1. Trough concentrations are only represented for infusions 2–6. Infusion 7 was the last dose. Dose groups ranged from ≤0.5 mg/kg to 24 mg/kg. Arrows indicate when a dose of daratumumab was administered. DARA daratumumab
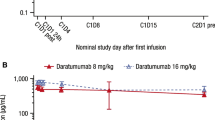
In Part 2 of the GEN501 study, the mean ± SD C max after the first full infusion of daratumumab in patients receiving 16 mg/kg (263.41 ± 92.00 μg/mL; n = 40) was two-fold higher than in patients receiving 8 mg/kg (133.54 ± 32.45 μg/mL; n = 28; Table 2). There was evidence to indicate accumulation of daratumumab, with mean C max increasing by 1.8 to 3.5 times from the first to the last full infusions. Calculation of t ½ was only possible for the 16 mg/kg dose group, with considerable variability. Similar to Part 1 of the study, clearance was higher after the first full infusion than after the last infusion. Following the first administration at the recommended dose of 16 mg/kg, mean clearance was 0.42 ± 0.42 mL/h/kg and, after repeat administration, was decreased to 0.30 ± 0.12 mL/h/kg. The mean daratumumab concentration–time profile following the first 16 mg/kg infusion is presented in Fig. 3.

Mean daratumumab serum concentration in Part 2 of the GEN501 study following the first full infusion. Error bars represent mean ± 1.96 standard error (95% confidence interval). DARA daratumumab, EOI end of infusion
3.2 SIRIUS Study
In the SIRIUS study, the mean concentration of daratumumab at the end of the first infusion was approximately 2.3-fold higher in the 16 mg/kg group (312.54 μg/mL) than in the 8 mg/kg group (137.55 μg/mL), which was approximately dose proportional.
After the first intravenous infusion of daratumumab 8 mg/kg, mean serum concentrations peaked at the EOI, then declined in an apparent bi-exponential manner, and remained quantifiable through 28 days post-dose (second infusion pre-dose). Following the last full infusion, serum daratumumab concentrations fell below the limit of quantification by 8 weeks post-treatment in the 8 mg/kg group, but remained detectable in most patients in the 16 mg/kg group at this timepoint.
In the daratumumab 16 mg/kg group, there was evidence of drug accumulation through the first two cycles; mean ± SD trough concentration at the end of weekly dosing was 573.49 ± 331.49 μg/mL, rising to 914.86 ± 410.34 μg/mL at the end of the infusion (ninth planned infusion), with the latter being approximately 2.9-fold greater than at the end of the first infusion (312.54 ± 106.65 μg/mL). After patients entered cycle 3, when dosing frequency decreased to every 2 weeks, peak and trough serum daratumumab concentrations decreased slightly, with mean trough concentrations maintained above 400 μg/mL through cycle 6.
Pharmacokinetic parameters were only derived for patients receiving daratumumab 8 mg/kg due to the drug administration and sampling schedules (Table 3). Following the first dose of daratumumab 8 mg/kg, the mean ± SD apparent t ½ was 120.69 ± 105.818 h, mean ± SD clearance was 0.52 ± 0.253 mL/h/kg, and mean ± SD V Z was 61.99 ± 18.425 mL/kg. The coefficient of variation (CV%) was approximately 64% for AUCinf values and approximately 88% for t ½, demonstrating a large interpatient variability in systemic exposure to daratumumab after the first 8 mg/kg dose, which may be reflective of the variable impact of target-mediated drug disposition at this dose level.
3.3 Correlation Between Daratumumab Disposition and Reduction in Paraprotein
The majority of patients exhibited a decrease in paraprotein from baseline. In GEN501, 80% of patients receiving daratumumab 16 mg/kg had a reduction in paraprotein; 46 and 24% of patients had a >50 and >90% reduction, respectively. Among the patients who were administered daratumumab 16 mg/kg in the SIRIUS study, 38 and 16% of patients had a >50 and >90% reduction in paraprotein from baseline, respectively. There was a significant positive correlation between maximum reduction in paraprotein and daratumumab exposure (Fig. 4). Maximum reduction in paraprotein appeared to plateau when daratumumab exposure was high; maximum reduction in paraprotein showed the strongest correlation with maximal pre-infusion concentration.

Maximal reduction in paraprotein from baseline versus maximal pre-infusion concentration in the GEN501 and SIRIUS studies. The solid blue line represents the smoothing curve of the data and the gray band represents the 95% confidence interval of the smoother. Pooled data from the GEN501 and SIRIUS studies are shown
4 Discussion
Daratumumab exhibited typical pharmacokinetics for an IgG1 monoclonal antibody; mean serum concentrations of daratumumab peaked at the end of the first infusion and then declined in an apparent bi-exponential manner. V Z estimates indicate that daratumumab is confined primarily in the vascular system, with limited extravascular tissue distribution. Following the first full infusion, C max increased in approximate proportion to the increasing dose of daratumumab 1–24 mg/kg, and increased in a greater than dose-proportional manner after multiple doses. AUC increased in a greater than dose-proportional manner after the first and multiple doses. Following the 16 mg/kg recommended dosing regimen (weekly for 8 weeks, every 2 weeks for 16 weeks, and every 4 weeks thereafter), accumulation of daratumumab continued throughout the weekly dosing period and decreased slightly as patients entered the less intense dosing periods. Following the end of treatment, daratumumab remained detectable in most patients in the SIRIUS 16 mg/kg group at 8 weeks post-treatment. No subjects in the GEN501 or SIRIUS studies developed antibodies to daratumumab during treatment [15, 16], suggesting a low risk for immunogenicity with this therapeutic agent.
Decreasing clearance with increasing dose levels was observed consistently across both studies, and data from the GEN501 clinical trial showed that the clearance also decreased with repeated dosing within the same dose level. As expected with the changes in clearance, the mean t½ also increased with increasing dose and repeated dose administration. These data demonstrate that daratumumab exhibits nonlinear pharmacokinetic characteristics, which is consistent with the target-mediated drug disposition of daratumumab observed in a recent study (Xu et al., submitted). Decreasing daratumumab clearance with increasing dose levels may indicate saturation of target-mediated clearance at higher dose levels, whereas decreasing clearance over time with repeated dosing may be due to a reduction of tumor burden as CD38+ cells are eliminated. Similarly, another IgG monoclonal antibody approved for the treatment of MM, elotuzumab, demonstrated nonlinear pharmacokinetics consistent with target-mediated clearance when administered in combination with lenalidomide and dexamethasone [18].
Following the first full dose of daratumumab, data from the SIRIUS study showed larger interpatient variability in exposure and t ½ at the 8 mg/kg dose level (88% and 64% CV for t ½ and AUC, respectively, in Parts 1 and 2) compared with the 16 mg/kg dose level in GEN501 (38–48% CV for t ½ and AUC in Parts 1 and 2). This may reflect the variable impact of target-mediated drug disposition, in which drug may saturate the target in most patients. This observation is consistent with exposure-response analyses described elsewhere (Xu et al, submitted), which indicated that most patients (approximately 80%) following the recommended 16 mg/kg dose regimen were expected to achieve daratumumab serum concentrations correlated with 99% target (CD38) saturation and 90% of the maximal effect on ORR. At the lower 8 mg/kg dose level, and following the same dose schedule, only 40–50% of patients are expected to achieve concentrations related to these target saturation and ORR thresholds. Consistent with these findings, patients treated with daratumumab 8 mg/kg demonstrated a lower ORR compared with those treated with daratumumab 16 mg/kg in the GEN501 (10 vs. 36%, respectively) [15] and SIRIUS studies (11 vs. 29%, respectively) [16].
There are some intrinsic limitations of these analyses due to the study designs of GEN501 and SIRIUS, as phase I/II and phase II studies, respectively. The patient numbers were relatively small and analyses of pharmacokinetics over time were limited by the schedule for pharmacokinetic sampling defined by the protocol of each study. As a result, for some dosing schedules, few samples that met the criteria for pharmacokinetic analysis were available. Despite these limitations, the pharmacokinetic data consistently indicate that daratumumab has nonlinear pharmacokinetics consistent with target-mediated drug disposition.
5 Conclusions
The data reported here, in conjunction with other analyses, support a dose regimen of daratumumab 16 mg/kg weekly for 8 weeks, every 2 weeks for 16 weeks, and every 4 weeks thereafter, which rapidly saturates target-mediated clearance during weekly dosing and maintains saturation when dosing occurs every 2 or 4 weeks [19]. Daratumumab is approved in the US, Europe, and Canada for the treatment of heavily pretreated MM patients, and is administered at 16 mg/kg according to the dosing regimen described above [19–21].
References
Harousseau JL. Ten years of improvement in the management of multiple myeloma: 2000–2010. Clin Lymphoma Myeloma Leuk. 2010;10:424–42.
Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111:2516–20.
Kumar SK, Dispenzieri A, Lacy MQ, et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia. 2014;28:1122–8.
Cavo M, Rajkumar SV, Palumbo A, et al. International Myeloma Working Group consensus approach to the treatment of multiple myeloma patients who are candidates for autologous stem cell transplantation. Blood. 2011;117:6063–73.
Palumbo A, Rajkumar SV, San Miguel JF, et al. International Myeloma Working Group consensus statement for the management, treatment, and supportive care of patients with myeloma not eligible for standard autologous stem-cell transplantation. J Clin Oncol. 2014;32:587–600.
Kumar SK, Lee JH, Lahuerta JJ, et al. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: a multicenter International Myeloma Working Group study. Leukemia. 2012;26:149–57.
Meadows JP, Mark TM. Management of double-refractory multiple myeloma. Curr Hematol Malig Rep. 2013;8:253–60.
van de Donk NW, Lokhorst HM. New developments in the management and treatment of newly diagnosed and relapsed/refractory multiple myeloma patients. Expert Opin Pharmacother. 2013;14:1569–73.
McKeage K. Daratumumab: first global approval. Drugs. 2016;76:275–81.
Lin P, Owens R, Tricot G, et al. Flow cytometric immunophenotypic analysis of 306 cases of multiple myeloma. Am J Clin Pathol. 2004;121:482–8.
Santonocito AM, Consoli U, Bagnato S, et al. Flow cytometric detection of aneuploid CD38(++) plasmacells and CD19(+) B-lymphocytes in bone marrow, peripheral blood and PBSC harvest in multiple myeloma patients. Leuk Res. 2004;28:469–77.
de Weers M, Tai YT, van der Veer MS, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol. 2011;186:1840–8.
Overdijk MB, Verploegen S, Bogels M, et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. MAbs. 2015;7:311–21.
Krejcik J, Casneuf T, Nijhof IS, et al. Daratumumab depletes CD38+ immune-regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood. 2016;128:384–94.
Lokhorst HM, Plesner T, Laubach JP, et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med. 2015;373:1207–19.
Lonial S, Weiss B, Usmani S, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet. 2016;387:1551–60.
US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research. Guidance for Industry. Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system. August 2000. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070246.pdf. Accessed 11 May 2011.
Empliciti (elotuzumab) for injection [package insert]. New York, NY: Bristol-Myers Squibb; 2015.
Darzalex™ (daratumumab) injection, for intravenous use [package insert]. Horsham, PA: Janssen Biotech, Inc.; 2015.
European Medicines Agency. EPAR summary for the public. Darzalex daratumumab. 2016. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/004077/WC500207298.pdf. Accessed 21 June 2016.
The Myeloma Beacon. Darzalex (daratumumab) approved by Health Canada as the first human anti-CD38 monoclonal antibody for the treatment of multiple myeloma. 2016. Available at: http://www.myelomabeacon.com/pr/2016/06/30/darzalex-approved-by-health-canada/. Accessed 9 Aug 2016.
Acknowledgements
The authors thank Qun (Trina) Jiao, MS; Xiang Alice Zong, MSPH; and Marijke Adams, PharmD, PhD, for contributions to the pharmacokinetic parameter analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was sponsored by Janssen Research & Development, LLC. Medical writing and editorial support was provided by Erica S. Chevalier-Larsen, PhD, of MedErgy and was funded by Janssen Global Services, LLC. Open access to this report was sponsored by Janssen Research & Development, LLC.
Conflict of interest/disclosure
Pamela L. Clemens, Xiaoyu Yan, Imran Khan, Richard Jansson, Tahamtan Ahmadi, Kristen Lantz, Honghui Zhou, Thomas Puchalski, and Xu Steven Xu are employees of Janssen Research & Development, LLC. Pamela L. Clemens, Richard Jansson, Kristen Lantz, Honghui Zhou, Thomas Puchalski, and Xu Steven Xu own stock in Johnson & Johnson. Henk M. Lokhorst reports honoraria from Amgen, and honoraria and research funding from Genmab and Janssen. Sagar Lonial reports consultancy and research funding from Millennium, Novartis, Bristol-Myers Squibb, Onyx, Celgene, and Janssen. Nedjad Losic is an employee of and owns stock in Genmab A/S.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Clemens, P.L., Yan, X., Lokhorst, H.M. et al. Pharmacokinetics of Daratumumab Following Intravenous Infusion in Relapsed or Refractory Multiple Myeloma After Prior Proteasome Inhibitor and Immunomodulatory Drug Treatment. Clin Pharmacokinet 56, 915–924 (2017). https://doi.org/10.1007/s40262-016-0477-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-016-0477-1