
Abstract
Necitumumab is a second-generation, recombinant, human immunoglobulin G1, epidermal growth factor (EGFR) receptor antibody that specifically blocks the ligand binding site of EGFR. Necitumumab potentially acts by blocking ligand epidermal growth factor (EGF) binding-mediated activation of the EGFR signaling pathway, inhibiting tumor growth, angiogenesis, and anti-apoptotic mechanisms. Necitumumab inhibited the interaction of EGF and EGFR with a concentration that inhibits binding by 50 % of approximately 0.9 nM (0.13 mg/L) and demonstrated antitumor activity during in vivo experiments associated with trough plasma concentrations of approximately 40 mg/L. This work describes the population pharmacokinetics of necitumumab in cancer patients when administered with or without concomitant chemotherapy and evaluates patient characteristics that may guide dosing. Nonlinear mixed-effects modeling of serum concentration data across five clinical studies (phases I–III) indicated that necitumumab exhibited target-mediated drug disposition, commonly observed with monoclonal antibodies, and that pharmacokinetics were expected to be linear in the studied dose ranges when administered as repeated infusions. No age, sex, race, or concomitant medication factors were found influential, while weight was a statistically significant factor for both distribution and elimination. Simulations from the final model indicated that only a limited reduction in patient drug exposure variability would be achieved by weight- or body surface area-based dosing. Necitumumab effective half-life was estimated to approximately 2 weeks, and steady state was achieved within three to four cycles of treatment. The phase III dosing schedule of 800 mg dosed on days 1 and 8 of a 21-day schedule resulted in serum concentrations that exceeded the 40-mg/L threshold indicated by preclinical experiments.
Similar content being viewed by others
Necitumumab pharmacokinetics is characterized by target-mediated drug disposition, with a volume of distribution approximate to blood volume and a half-life of 2 weeks. |
No age, sex, race, or concomitant medication factors were found influential, while weight explained a limited percentage of variability in distribution and elimination. |
Simulations from the final model indicated that a only minor reduction in patient drug exposure variability would be achieved by weight- or body surface area-based dosing. |
1 Background
Necitumumab (also named IMC-11F8) is a DNA-derived, second-generation, recombinant human immunoglobulin G1 (IgG1), anti-epidermal growth factor receptor (EGFR) monoclonal antibody (mAb) that specifically binds to the immobilized EGFR with high affinity to inhibit epidermal growth factor (EGF)-induced EGFR phosphorylation [1]. This antibody potently antagonizes the binding of the EGF ligand to EGFR, inhibits EGF-stimulated activation of both EGFR and the downstream p44/p42 MAP kinases, and neutralizes EGF-induced DiFi cell proliferation. Further to inhibiting ligand-induced EGFR activation, necitumumab was capable of inducing an antibody-dependent, cell-mediated cytotoxic response against DiFi cells by human peripheral blood mononuclear cells. Necitumumab inhibited the interaction of EGF and EGFR with a concentration that inhibits binding by 50 % of approximately 0.9 nM (0.13 mg/L). The nonclinical pharmacology package includes in vitro studies to characterize the binding and functional inhibition of the human EGFR by necitumumab. The in vivo antitumor activity of necitumumab was evaluated in colon, non-small-cell lung, pancreatic, and squamous cervical cancer xenograft tumor models in athymic mice. Necitumumab demonstrated significant antitumor activity as a monotherapy in colon, non-small-cell lung, pancreatic, and squamous cervical cancer models. Antitumor activity of necitumumab in preclinical in vivo experiments was observed down to doses of 4–6 mg/kg, twice per week, associated with trough plasma concentrations of approximately 40 mg/L. Overall, the preclinical pharmacodynamic data demonstrated that necitumumab has the potential to impact human cancer progression.
Pharmacokinetic (PK) results of non-compartmental analysis to date suggest that, as with other recombinant human (IgG1) mAbs targeting cell surface antigens [2], necitumumab exhibited nonlinear PK characteristics [3]. Apparent nonlinear PK profiles were observed between 100 and 1000 mg. Clearance (CL) after multiple doses was lower than that after a single dose. Following 7 weekly doses, the geometric mean accumulation ratio based on area under the curve ranged from 1.6 to 3.2. The maximum tolerated dose was determined to be 800 mg [3]. This work aims at presenting the population PK of necitumumab in cancer patients, and describing any influential patient factors determining drug exposure.
2 Methods
Population PK analyses of the data from these five studies were performed using NONMEM (Version 7.3, ICON Plc, Ireland). Serum concentrations below the quantification limit were treated as missing values and were not included in the population pharmacokinetics. In general, missing values of independent variables (demographic and laboratory) were imputed by the last observation carried forward method. Outlier testing was performed for necitumumab concentrations, according to the US Food and Drug Administration Guidance for Industry on Population Pharmacokinetics (1999) as well as internal standard operating procedures. Outlier testing for rich sample patient profiles were based on the ±3 standard deviations criteria, while sparse sample profiles are deemed at the discretion of the analyst, where only apparently unreasonable samples are omitted, such as high concentrations after several below the quantification limit values with no dose recorded in between, quantifiable pretreatment samples, and concentration measurements without corresponding dose information.
A series of one-, two-, and three-compartment models were evaluated to identify the model that best described the necitumumab concentration–time data. Additional model structures were evaluated as previous studies suggested that necitumumab exhibited an exposure-dependent disposition. An approximation of a target-mediated drug disposition model [4–6] and a Michaelis–Menten CL model were tested. Estimates of the PK parameters and error terms were obtained by fitting the concentration–time data by means of the nonlinear mixed-effects modeling program NONMEM (Version 7.3). The first-order conditional with interaction estimation method was used for all analyses (NONMEM User’s Guide, Part VII). The inter-patient variability was assumed to be log-normally distributed. Covariance between parameters was assessed using an omega block. Different residual error models—additional, proportional, and combined additive and proportional—were also evaluated.
Selection of the most appropriate PK base model was based upon agreement between predicted and observed serum concentrations, lack of pattern (that is, randomness) in the weighted residuals vs. the predicted values, non-positive average changes in the inter-patient variability, and significant decreases in the Minimum value of the objective function (MOF).
Parameter estimate precision was evaluated through bootstrapping on the final base model to confirm that all PK parameters were well estimated. A visual predictive check (VPC) was also performed on the base model to investigate the agreement between the observed and predicted concentrations.
Once a structural and statistical model was established, the effect of patient factors was assessed for their clinical relevance on the disposition of necitumumab. Covariate testing was performed without eta correlation. A listing of patient factors and the specific PK parameters on which they were tested are specified in Table 1.
Influence of patient bodyweight on CL and volume parameters was tested and incorporated as a power function (Eq. 2) with an inclusion criteria of p < 0.001. Simple allometric scaling was also tested, that is, a fixed power coefficient of 0.75 for CL parameters and 1 for volume of distribution parameters [7, 8]. Inclusion of fixed power coefficients does not infer any loss in degrees of freedom; therefore, the ΔMOF significance criteria are set aside and any drop of MOF corresponds to model improvement. Lean body mass and body surface area (BSA) were also investigated.
Next, stepwise covariate modeling (SCM) was implemented using Perl-Speaks NONMEM (PsN) [9, 10] for remaining demographic, liver function, disease state, and concomitant medication covariates. The criterion for forward inclusion was a p value no greater than 0.01 (Δ6.635 MOF for inclusion of one parameter) with a backward deletion threshold of 0.001 (Δ10.828 MOF) for exclusion of one parameter).
Continuous covariates were tested using linear, power, and exponential models as shown in Eqs. 1–3. Categorical covariates were tested using a categorical model, as shown in Eq. 4.
Here, P is the individual’s estimate of the parameter (for example, CL, V), θ 1 represents the typical value of the parameter, θ 2 represents the effect of the covariate, COV is the value of the covariate, and MED is the population median of the covariate. IND is an indicator variable with a value of either 0 or 1 assigned for values of a dichotomous categorical covariate (for example, female or male) and 1 for various values of a categorical covariate ranging from 1 to n, where n is the number of categories (for example, n races).
The final backward model from SCM was further reduced in a step-wise manner. If removal of a covariate did not result in a significant (p < 0.001) MOF drop and reduce IIV by 5 %, the covariate was removed.
Because sample age stability validation at the time of the analysis only encompassed 24 months, with 14 % of samples being between 24 and 36 month of storage age, samples were flagged as above or below 24 months of age. The effect of assay age on each of the residual error terms was tested as a dichotomous categorical covariate on the residual error parameters using significance criteria above (p < 0.001), and finally covariance was again assessed between CL and volume parameters using an omega block.
Final model development was based on statistical significance criterias, such as a ≥10.828 point drop in MOF (p < 0.001) and a decrease in the inter-subject variability in the relevant parameters of >5 %, as well as clinical relevance criteria, such as a covariate parameter effect being a >15 % difference for a dichotomous covariate or a >15 % covariate effect at the highest or lowest observed value for a continuous variable. In addition, goodness of fit was evaluated with residual plots and VPCs. Prediction correction of VPCs was applied where appropriate [11], to allow a comparison of model performance across dose concentrations and varying schedules. Bootstrap analysis with 1000 samples was performed to assess the precision of the final parameter estimates of base and final models, and to calculate the 95 % confidence interval for all parameters.
To evaluate the impact of the selected flat dose paradigm, simulations from the final model were performed, evaluating the distribution of predicted C ss,ave with the 800-mg dose compared with alternate weight or BSA-based dosing paradigms, assuming the same weight distribution in the phase III study SQUIRE, i.e., a median (range) of 70 (35–127) kg. The weight- and BSA-based dose concentrations were selected to produce a similar median C ss,ave as the flat dose, and this measure was chosen for comparison as it reflects the overall drug exposure. Therefore, based on each patient’s individual PK parameters (474 patients in total) from the final model, C ss,ave values were predicted based on a flat dose, or a 11-mg/kg dose or 450 mg/m2 and compared with the same patient receiving 800 mg.
2.1 Dosing
Necitumumab has been administered to humans using several different dosing regimens. For the present analysis, data were pooled from studies where necitumumab was administered at two dose concentrations (600 and 800 mg) and with different dose intervals (weekly, biweekly, or days 1 and 8 of a 3-week cycle), given as infusions over approximately 1 h. Necitumumab was administered both as a single agent and in combination with several chemotherapeutic drugs (cisplatin, pemetrexed, and gemcitabine) and to patients with several tumor types [3, 12–14].
2.2 Study Population
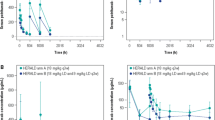
The population PK evaluation included 4920 necitumumab concentrations from 807 patients across five studies, with data available from 15 to 407 patients (Figs. 1, 2). Three phase I/II studies included rich PK sampling in patients with advanced solid cancers, while two phase III studies included sparse sampling (trough only) in squamous and non-squamous non-small-cell lung cancer patients [3, 12–14]. Continuous and categorical characteristics for these patients are summarized in Tables 2 and 3. One hundred and eighty-four samples, drawn from a total of 123 patients, were non-quantifiable owing to concentrations below the quantitation limit of assay. No outlier patients were identified and omitted from the analysis dataset according to the standard operating procedure, however, eight necitumumab concentrations from one patient were excluded from the analysis, as they were inconsistent with the PK properties of the drug in that concentrations reported predose were higher than those reported at 1-h post-end of infusion or high concentrations were reported following a concentration below the quantitation limit with no additional dosing information recorded.

Distribution of sampling times relative to the beginning of the last necitumumab infusion

Observed necitumumab concentration vs. time from the start of the first necitumumab infusion
3 Results
The necitumumab concentration–time data were best described by a two-compartment structural model parameterized in terms of central volume of distribution (V 1), peripheral volume of distribution (V 2), and inter-compartmental CL (Q). Drug CL was best described by an approximation of the target-mediated drug disposition model [4–6], using [1] a saturable component parameterized according to Michaelis–Menten (K m and V max) and [2] first-order CL describing non-target-mediated degradation, both acting in the central compartment. Exponential inter-patient variability terms were included for total drug CL (CLtot), V 1, and V 2. Residual variability was accounted for by an additive/proportional error structure. The population PK parameters CL, V 1, V 2, and Q were well estimated, with relative standard error (RSE) at or below 6.1 %, while the Michaelis–Menten parameters K m and V max had higher uncertainty (Table 4).
Once the base structural model was established, potentially significant covariates were evaluated as described. Estimation of power coefficients for bodyweight resulted in a better fit to data than fixed power allometry, and was retained through the covariate search. The resulting covariate model after forward inclusion and backward deletion from the SCM, using total patient bodyweight or lean body mass performed very similarly (ΔMOF = 2.54); therefore, total bodyweight was selected as being a more practical measure for clinical purposes.
After application of pre-specified inclusion/exclusion criteria, only patient bodyweight remained as a significant covariate for CL and volume parameters. Parameters were estimated to be less than proportionally dependent on weight; for CL parameters, the power coefficient was estimated at 0.768 and for volume parameters it was 0.498. The population PK parameters CL, K m, V max, V 1, V 2, and Q were well estimated, as were the covariate bodyweight effects. Final parameter estimates are shown in Table 4.
The estimated inter-patient variability for CLtot, V 1, and V 2 was significantly decreased compared with the base model. The interaction between CLtot and V 1 was well characterized. CLtot is the sum of linear and nonlinear clearances [CLtot = CL + V max/(C + K m)]. Simulations were used to calculate the population estimates of V ss (6.97 L) and CLtot parameters (0.0141 L/h) at steady state following the phase III study dosing with 800 mg on days 1 and 8 of a 21-day cycle [CLtot,ss = CL + V max/(C ss,ave + K m), V ss = V 1 + V 2].
Goodness of fit for the final population model is represented graphically (Fig. 3) by the agreement between predicted values (Population prediction (PRED) and Individual Prediction (IPRED)) and observed concentrations (Dependent value (DV)), as well as the randomness of the individual and conditional weighted residuals. In general, there was a random distribution of residual values suggesting no consistent bias in the predicted necitumumab concentrations, as further demonstrated by the VPC (see Fig. 4).

Predicted concentrations and weighted residuals for the necitumumab (a) base model and the (b) final model. Conc concentration

Prediction-corrected visual predictive check of the final pharmacokinetic model. Blue circles are observed data. The solid line depicts median observed data, while the pink shaded area defines the prediction interval around the median. The dashed line depicts the observed 5th and 95th percentiles, while the blue shaded area defines the prediction interval of the same
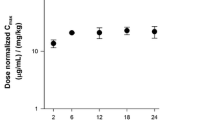
Simulations from the final model evaluated the distribution of predicted C ss,ave, resulting from a 800-mg flat dose compared with a weight- or BSA-based dosing paradigm using 11.5 mg/kg or 450 mg/m2 respectively, with results shown in Fig. 5.

Predicted Css,ave necitumumab concentrations based on a flat 800-mg, weight-based (11.5 mg/kg), and body surface area-based (450 mg/m2) dose regimen administered on days 1 and 8 of a 3-week regimen
4 Discussion
It is well known that PK properties of mAbs differ significantly from those of small-molecule drugs. Disposition of mAbs is usually influenced by target-related factors including target antigen expression level and turnover rate, immune reactions to the antibody, target antigen distribution, and binding affinity [2, 15]. In solid tumors, the distribution may depend on the architecture and physiology of the tumors [16, 17].
The PK parameters of necitumumab were well characterized by an approximation of the target-mediated drug disposition model [4–6]. The model contained a central and a peripheral volume of distribution, and elimination was characterized by a linear and a saturable component, assumed to reflect binding to target. The population estimates of V ss (6.97 L) and CLtot parameters (0.0141 L/h) at steady state following dosing of 800 mg on days 1 and 8 of a 21-day cycle were in range with other human IgG mAbs studied in the clinic [2, 16, 17], and translated into a functional half-life of approximately 2 weeks, with steady state achieved after three to four cycles of treatment.
The non-linear CL term K m was estimated to 7.97 mg/L, which is well exceeded with continuous treatment in the studied dose range. This means that linear kinetics could be assumed under normal clinical conditions, which leads to predictable effects of potential dose adjustments.
Effects of different covariates (including sex, age, bodyweight, race, albumin, hepatic function, and renal function) were investigated for influence on the disposition of necitumumab in the population PK analysis. Patient bodyweight was the only significant covariate on the necitumumab disposition that was retained in the final model, with a less than proportional effect on both CL and distribution parameters. The influence of bodyweight on steady state was moderate (typical CLtot ranged from 77 to 131 % and typical V ss from 84 to 120 % of the median at the 5th and 95th weight percentiles) in comparison to overall variability. Inter-patient variability in the PK parameters was moderate to high (28.8 % for CL, 21.1 % for V 1, and 55.4 % for V 2) as commonly observed with mAbs [18]. Inter-occasion variability was not assessed owing to computational issues, as expected given studies with rich sampling only contained information from a single-dose administration, while long-term studies encompassed only trough sampling. The lack of correlation between liver or renal status and CLtot can be expected given the known mechanisms involved for the CL of antibodies, while a relationship to patient bodyweight could be expected [16–18]. Even though inclusion of patient bodyweight as a covariate fulfilled all statistical criteria, comparison of base and final model goodness-of-fit plots (Fig. 3) clearly indicates the overall contribution to variability from bodyweight is limited. The simulations comparing a flat dose- vs. a bodyweight-adjusted dose show that weight- or BSA-based dosing would not significantly decrease the overall variability in exposure, but rather increase the risk of dosing and administration errors, and why a flat dose paradigm would be preferred. During the clinical development of necitumumab, a target concentration of 40 mg/L has been used, as derived from the concentration needed to achieve tumor growth inhibition in in vivo xenograft experiments. The work presented here can be used to support the use of a flat dose in clinical development, but future work is needed to establish the exposure response of necitumumab, and whether the target concentration desired is representative of clinical anticancer activity, and whether the proposed dose intensity is sufficient to achieve efficacy.
5 Conclusions
Nonlinear mixed-effects modeling of serum concentration data across five clinical studies in various development phases indicate that necitumumab exhibits target-mediated drug disposition, as commonly observed with mAbs, and that pharmacokinetics can be expected to be linear in the studied dose ranges when administered as repeated infusions. No age, sex, race, or concomitant medication factors were found influential, while weight was a statistically significant factor for both distribution and elimination. Simulations from the final model indicate that only a limited reduction in patient drug exposure would be achieved by weight-or BSA-based dosing. The necitumumab effective half-life was estimated to approximately 2 weeks, and steady state was achieved within three to four cycles of treatment. The phase III dosing schedule of 800 mg dosed on days 1 and 8 of a 21-day schedule resulted in serum concentrations well exceeding the 40-mg/L threshold indicated by preclinical experiments.
References
Dienstmann R, Tabernero J. Necitumumab, a fully human IgG1 mAb directed against the EGFR for the potential treatment of cancer. Curr Opin Invest Drugs. 2010;11(12):1434–41.
Keizer RJ, Huitema AD, Schellens JH, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(8):493–507.
Kuenen B, Witteveen PO, Ruijter R, et al. A phase I pharmacologic study of necitumumab (IMC-11F8), a fully human IgG1 monoclonal antibody directed against EGFR in patients with advanced solid malignancies. Clin Cancer Res. 2010;16(6):1915–23.
Gibiansky L, Gibiansky E. Target-mediated drug disposition model: approximations, identifiability of model parameters and applications to the population pharmacokinetic-pharmacodynamic modeling of biologics. Exp Opin Drug Metab Toxicol. 2009;5(7):803–12.
Gibiansky L, Gibiansky E. Target-mediated drug disposition model and its approximations for antibody-drug conjugates. J Pharmacokinet Pharmacodynam. 2014;41(1):35–47.
Gibiansky L, Gibiansky E, Kakkar T, Ma P. Approximations of the target-mediated drug disposition model and identifiability of model parameters. J Pharmacokinet Pharmacodyn. 2008;35(5):573–91.
Holford NH. A size standard for pharmacokinetics. Clin Pharmacokinet. 1996;30(5):329–32.
Holford N, Heo YA, Anderson B. A pharmacokinetic standard for babies and adults. J Pharm Sci. 2013;102(9):2941–52.
Lindbom L, Pihlgren P, Jonsson EN. PsN-Toolkit: a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Progr Biomed. 2005;79(3):241–57.
Lindbom L, Ribbing J, Jonsson EN. Perl-speaks-NONMEM (PsN): a Perl module for NONMEM related programming. Comput Methods Progr Biomed. 2004;75(2):85–94.
Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2006;13(2):143–51.
Elez E, Hendlisz A, Delaunoit T, Sastre J, et al. Phase II study of necitumumab plus modified FOLFOX6 as first-line treatment in patients with locally advanced or metastatic colorectal cancer. Br J Cancer. 2016;114(4):372–80.
Paz-Ares L, Mezger J, Ciuleanu TE, et al. Necitumumab plus pemetrexed and cisplatin as first-line therapy in patients with stage IV non-squamous non-small-cell lung cancer (INSPIRE): an open-label, randomised, controlled phase 3 study. Lancet Oncol. 2015;16(3):328–37.
Thatcher N, Hirsch FR, Luft AV, et al. Necitumumab plus gemcitabine and cisplatin versus gemcitabine and cisplatin alone as first-line therapy in patients with stage IV squamous non-small-cell lung cancer (SQUIRE): an open-label, randomised, controlled phase 3 trial. Lancet Oncol. 2015;16(7):763–74.
Deng R, Iyer S, Theil FP, et al. Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: what have we learned? mAbs. 2011;3(1):61–6.
Dostalek M, Gardner I, Gurbaxani BM, et al. Pharmacokinetics, pharmacodynamics and physiologically-based pharmacokinetic modelling of monoclonal antibodies. Clin Pharmacokinet. 2013;52(2):83–124.
Tabrizi M, Funelas C, Suria H. Application of quantitative pharmacology in development of therapeutic monoclonal antibodies. AAPS J. 2010;12(4):592–7601.
Dirks NL, Nolting A, Kovar A, Meibohm B. Population pharmacokinetics of cetuximab in patients with squamous cell carcinoma of the head and neck. J Clin Pharmacol. 2008;48(3):267–78.
Acknowledgments
The authors are indebted to Michael Heathman for expert input and modeling advice.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The studies used in this work were conducted in compliance with the Declaration of Helsinki, International Conference on Harmonization Guidelines for Good Clinical Practice, and applicable local regulations. The protocol was approved by the ethics committees of all participating centers and all patients provided written informed consent before study entry.
Funding
The authors are employed by Eli Lilly & Co, and all work presented was funded by the company.
Conflict of interest
Amanda Long, Emmanuel Chigutsa and Johan Wallin are all employees of Eli Lilly & Co, and hold stock options or shares in the company.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Long, A., Chigutsa, E. & Wallin, J. Population Pharmacokinetics of Necitumumab in Cancer Patients. Clin Pharmacokinet 56, 505–514 (2017). https://doi.org/10.1007/s40262-016-0452-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-016-0452-x