
Abstract
Following the publication of our paper regarding a population-based model of doxorubicin pharmacokinetics in children in Clinical Pharmacokinetics last year (Voller et al. 54:1139–1149, 2015), we have received many inquiries on the practical clinical consequences of this model; however, a population-based model is only one of the aspects to be taken into account when developing dosing algorithms. In addition, any new method of dose calculation would need clinical validation and, subsequently, a new clinical trial. However, such a trial, especially with regard to burden to the children involved, requires optimal preparation and the selection of the best hypotheses. The European Paediatric Oncology Off-Patent Medicines Consortium (EPOC), represented by the authors, would therefore like to initiate an interdisciplinary discussion on the clinical and pharmacological goals for dose calculation. This current opinion summarizes the existing knowledge on the pharmacokinetics and pharmacodynamics of doxorubicin. Our aim was to define the clinical needs as precisely as possible, with the intention of stimulating discussion between the clinical pediatric oncologist and the pediatric pharmacologist. By doing so, we hope to define surrogates for best practice of a common doxorubicin dose in children. The intent is for a trial to validate a rational dose calculation rule, leading to a regulatory process and subsequent labeling.
Similar content being viewed by others


New data on the pharmacokinetics of doxorubicin in children (0–18 years) have become available. |
Current dosing algorithms show a high variability. |
A well-defined target pharmacokinetic or pharmacodynamic parameter for doxorubicin is lacking. |
Consensus between different treatment groups is desirable to develop and validate safe and efficacious dosing guidelines. |
1 Introduction
The European Medicines Agency included doxorubicin on the ‘priority list’ (doc. ref. EMEA/197972/2007, London, June 2007) for medications with a high priority for further research on pediatric use, with the absence of pharmacokinetic knowledge being the most critical point. Based on this document, the European Paediatric Oncology Off-Patent Medicines Consortium (EPOC) set up a trial to investigate the pharmacokinetics of doxorubicin in children. The pharmacokinetic phase-II-trial (EudraCT number 2009-011454-17, short title: EPOC) was funded under the European Commission’s Seventh Framework Program, grant agreement number 222910. These regulatory and political activities had the objective of including more detailed dosing information in the summary of product characteristics and the package leaflets.
Doxorubicin is currently authorized for a number of malignancies in children, such as acute lymphoblastic leukemia (ALL), acute myeloid leukemia, Wilms’ tumor, Ewing’s sarcoma, osteosarcoma, and soft tissue sarcoma. Anthracyclines, such as doxorubicin, are currently used in nearly 60 % of children diagnosed with cancer [2], with a high impact on therapeutic success. Anthracyclines significantly increase event-free survival in patients with Ewing’s sarcoma, and are considered fundamental in the treatment of lymphomas and many solid tumors (i.e. soft tissue sarcomas, high-risk hepatoblastoma, and high-risk renal tumors) [2, 3]. There is also a non-significant tendency towards greater antitumor efficacy of anthracyclines in children diagnosed with ALL [2].
However, the most threatening drawback of this drug class is its dose-dependent late cardiotoxicity. Long-term survivors of childhood cancer have approximately five- to sixfold greater risk of cardiac dysfunction compared with their healthy siblings [4]. When observing the same cohort beyond the age of 35 years, the risk is increased by seven- to eightfold [5], which highlights the progressively increasing risk of cardiotoxicity with time.
The incidence of cardiotoxicity is clearly associated with cumulative dose, with doses greater than 300 mg/m2 resulting in a higher risk of cardiotoxicity [6]. Nevertheless, subclinical cardiotoxicity is already present at lower doses [7]. Generally, cumulative dose as a surrogate for ‘applied drug exposure’Footnote 1 is an accepted biomarker for cardiac damage. Because younger age at diagnosis, particularly below 4 years, has been associated with an inferior cardiac outcome [8], dose reduction in the very young is mandated in virtually all treatment protocols, following a long-established general principle in pediatric oncology [9]. Nonetheless, the influence of doxorubicin pharmacokinetics on both antitumor effect and cardiotoxicity remains unclear. The impact of a reduced dose on in vivo dose intensity in children is therefore largely unknown. Furthermore, high variability in the incidence of cardiotoxicity, even after accounting for clinical risk factors, might suggest an underlying genetic mechanism. Several candidate genes have been identified but, to date, no genes have impacted dosing guidelines [10].
In the past, it has been unclear whether drug elimination, quantified as clearance (CL), is lower in the very young than in older children. While one study indicated that CL is lower [11], others did not observe this tendency [12–14]. Data of our recent trial of doxorubicin in 101 children clearly prove that CL (L/h/m2), corrected for body surface area (BSA), is considerably lower in younger children [1]. Based on this new information, we ask the question as to how a rational dosing of doxorubicin in children can be achieved.
In order to stimulate this discussion, we visualize the effects of common dose adaptations on hypothetical children and real-life patients who participated in the trial [1]. Using the data of the population pharmacokinetic model developed for the EPOC trial, we consider how dosing guidelines based on pharmacokinetic data could be designed in children.
2 Current Dosing Concepts in Children
In 1958, Pinkel et al. postulated, using methotrexate as an example, that BSA should be the factor by which dosage in anticancer treatment should be adapted [15]. Based on the aim of reducing interindividual variability, BSA-based dosing became the standard approach in pediatric oncology [9].
However, in some cases, dosing in infants is performed based on body weight instead of BSA (with 30 kg ≙ 1 m2). The ratio of BSA to body weight in neonates and very young children far exceeds that of older children and adults due to physiological and anatomical development [16]. Thus, body-weight-based dosing in the youngest age group results in lower doses and thereby lower exposure.
Data in adults suggest that obese patients might have a more than twofold increase in exposure to doxorubicin when administered based on BSA [17]. Thus, even though no data in children are available, dosing based on BSA might not be suitable for the constantly increasing subgroup of morbidly obese patients.
An overview of commonly applied dose modifications, based on BSA, body weight or a combination thereof, is shown in Table 1. In case of BSA-based dosing, there is inconsistency in the extent of dose reduction, e.g. a 33 % reduction of the BSA-adjusted dose in children younger than 7 months in one protocol, and a 50 % reduction in the same population in another protocol.
In the case of weight-based dose adaptation, the weight cut-off is highly variable. One protocol reduces the dose in children up to 1 year of age or less than 12 kg, while others recommend an additional reduction of the body-weight-based dose by one-third in children younger than 7 months or weighing less than 5 kg.
This brief overview highlights the variability and arbitrary nature of dose adaptations, directly leading to an impact on drug exposure and dose intensity.
3 Evaluation of the Status Quo
3.1 Impact of Different Dose Recommendations on Hypothetical Children from Different Percentiles of Height and Body Weight
In order to study the impact of age and body weight on different dosing algorithms, three children with different body compositions were simulated. One child was assumed to be in the 5th percentile, one child in the 50th percentile, and one child in the 95th percentile of height and body weight from birth to adulthood [18]. The influence of the dose adaptations, presented in Table 1, on these three patients was evaluated.
For comparison, a ratio was calculated based on the protocol-based dose divided by a reference dose. The reference dose was defined as a dose adjusted only to the actual BSA of the patient, while the protocol-based dose was as specified in the various protocols presented in Table 1 (Eq. 1).
Inconsistencies in current protocols led to a difference in the percentage of BSA dose of up to one-third (Fig. 1). Besides the extent of dose reduction, the cut-off for termination of dose reduction is highly variable. Dose reductions up to 1 year are included in all studied protocols (Fig. 1); however, contrary to age-based cut-offs (protocol A: ALL-BFM; protocol C: CWS-2002, CWSSoTiSaR), body weight cut-offs are highly dependent on the body composition of the child. Differences were most obvious in the small and low-weight child (Fig. 1, solid lines), who reaches the 10 kg cut-off (protocol D: NB 2004/STS 2006) at approximately 2 years of age, and the 12 kg cut-off (protocol B: SIOP WT 2001; protocol E: SIOPEN HR-NBL-1) at approximately 3 years of age. In the child with a typical body weight and height (Fig. 1, dotted lines) the 10 kg cut-off is reached at an age marginally above 1 year, and the 12 kg cut-off is reached at an age of approximately 2 years. With regard to the tall and obese child, only a small difference in the maximal age of dose reduction was observed (all 1 year, or marginally above 1 year) (Fig. 1, dashed lines).

Effect of different dose adjustment schemes on the relative proportion of BSA dose administered to hypothetical patients on the 5th (solid line), 50th (dotted line) and 95th (dashed line) percentile of height and body weight. a protocol A; b protocol B; c protocol C; d protocol D; e protocol E according to Table 1; and f the dose adjustment based on the population pharmacokinetic model (in case no dose-adjustment based on body weight was recommended by the protocol, only one solid line is present). BSA body surface area
Based on this evaluation, clinicians should ask themselves whether they want to accept differences of up to one-third when dosing the patient according to one protocol or another. Another question to consider would be whether the maturation of a child can be adequately reflected by such discrete steps in a dosing algorithm.
3.2 Impact of Different Dose Recommendations on the Affected Patient Cohort of the EPOC-MS-001-DOXO Trial
The EPOC trial was performed in order to systematically investigate the pharmacokinetics of doxorubicin in children. Overall, 101 children (range 0.2–17.7 years; <3 years, n = 27), treated according to their tumor-specific protocols, were recruited [1]. The influence of dose adaptations in different protocols (Table 1) was evaluated in patients in the EPOC study specifically affected by protocol differences (age ≥1 year, body weight <12 kg). The analysis was carried out using the actual BSA, body weight and age of the patients.
The EPOC population included a number of children who were anomalously affected by the body-weight-based cut-offs: four children were older than 1 year of age, but below 10 kg (1.38, 1.42, 1.43 and 1.68 years), and six children were older than 1 year of age and between 10 and 12 kg (1.04, 1.59, 1.94, 2.29, 2.53 and 4.15 years). Thus, 9 of 19 children between 1 and 3 years of age, and one child above 3 years of age, would receive different doses depending on the protocol, resulting in pronounced differences in the administered percentage of BSA dose (Table 2). For example, the child aged 2.53 years, weighing 11.9 kg, would receive 66 % of the reference dose based on the BSA of protocol B (SIOP WT 2001), 75 % of the dose based on protocol E (SIOPEN HR-NBL-1), and a full BSA dose when enrolled in any of the other three treatment protocols (Table 2).
Although body weight, including outliers, was evenly distributed in children younger than 5 years of age (20 patients below and 23 patients above the 50th percentile of body weight), the data of the EPOC trial underline that typical pediatric cancer populations include a broad variability of individuals, including outliers. Five of 43 children younger than 5 years of age were below the 5th percentile of height and body weight (Table 2). In such specific cases, a multidisciplinary consensus on dosing options should be considered, informed by the considerations presented in this article.
4 What Should We Aim at When Developing Pharmacokinetic-Guided Dosing?
Unlike busulfan [19], methotrexate [20] and carboplatin [21], no target parameter, such as area under the concentration–time curve (AUC), maximal plasma concentration (C max), minimal plasma concentration (C min) or time-over-threshold concentration, has been defined for anthracyclines. In contrast, the effect of pharmacokinetic parameters on toxicity has been studied. It is widely accepted that a higher exposure to the drug, as represented by a higher cumulative dose, is associated with a higher risk of cardiac damage [22, 23], but the question of whether a high C max is associated with a greater risk of cardiac injury is controversial.
Some publications imply that higher C max is associated with significantly higher rates of congestive heart failure (CHF) when applying the same dose [24]. One investigation including 3184 children and comparing a 3-weekly schedule of doxorubicin with a weekly schedule (mean cumulative dose 240 mg/m2) found a 2.9 and 0.8 % rate of CHF in the 3-weekly and weekly dose groups, respectively [25]. However, another large study in children with ALL did not find a significant difference in cardiac function when comparing 1 h and 48 h infusions [26]. Since the most appropriate duration of infusion is unknown, there must be a balance between the risk of cardiotoxicity and patient convenience. Obviously, the dose administered must also be taken into account when selecting the duration of infusion.
As dose intensity is presumed to be the most relevant pharmacokinetic parameter for cytotoxic drugs, it might be reasonable to focus on the most commonly used measure of AUC. However, this approach does not directly consider the impact on other measures such as C max or time-over-threshold concentration, which have been intensively discussed for other drugs such as etoposide [27]. For a given infusion duration, these parameters vary proportionately. In addition, the optimal AUC for each treatment protocol depends on the other cytostatic drugs administered in combination with doxorubicin.
Taking the strategy of targeting AUC as an example, a further relevant question should be whether the aim is to achieve a uniform AUC across all patients. On the other hand, it could also be reasonable to target a lower AUC while maintaining efficacy, or even accepting lower efficacy in the very young as this is likely to be the population at highest risk for developing long-term toxicities such as CHF [8, 28]. Alternatively, a potentially higher AUC, and likely associated higher adverse event rate, could be accepted if a higher rate of cure resulted, as has been shown for acute myeloid leukemia [12].
No matter which parameter will be the focus of dosing, all parameters show wide intraindividual and interindividual variability. As the developed model shows considerable intraindividual variability on the central volume of distribution [1], a routine application of therapeutic drug monitoring would be unlikely to aid dose-finding in individual patients. A calculation rule has to therefore prioritize probabilities: not to exceed maximum AUCs/concentrations, not to fall short on minimums, and to fit for defined percentages of patients.
When attempting to develop dose adaptations, we also need to bear in mind that the number of children younger than 1 year of age studied to date is low. The EPOC trial is the largest and most recent trial of doxorubicin in children. Although, there was a specific focus on children less than 3 years of age, the EOPC trial included only four children younger than 1 year of age. The reasons for this lack of data are the rarity of oncologic diseases in this age group and the difficulty of recruiting such vulnerable subjects into clinical trials.
5 How Could We Attempt to Develop a Model-Based Dose Recommendation in Children?
In order to attempt to develop a dose adaptation, the population pharmacokinetic model of the EPOC trial, recently developed by our working group, was utilized [1]. The pharmacokinetics could be described by a three-compartment model in which all parameters were linearly scaled to BSA. In addition, the influence of age was modeled as a power function on CL.
Individual CL (CLi) was given using the following equation (Eq. 2):
where CLp represents the population estimate of CL, η i represents the deviation of the individual patient from the population value of CL (L/h), and BSAi (m2) and Agei (years) represent the individual age and BSA of each patient.
Based on this model, CL increases linearly with BSA; however, the model predicts an additional maturation of CL with age. CL (L/h/m2) increases rapidly in the very young, leveling off in older children (approximately 3–4 years). The predicted CL of a full-term newborn (BSA 0.22 m2) was 12.0 L/h/m2, while the model predicted a CL of 29.1 L/h/m2 in an 18-year-old (BSA 1.8 m2) [1].
During the development of the dose adaptation, it was assumed that a similar target drug exposure, calculated as AUC, should be attained in children of all ages. The AUC of an 18-year-old child (AUC18 years) was defined as the target AUC for all children, as maturation can be considered complete at that age and this approach might allow bridging to data available in adults.
Using Eq. 2, the nominal CL (L/h) was calculated for children of different ages and converted into the BSA-adjusted CL (L/h/m2). As the AUC is defined by the ratio of the applied dose and the individual patient’s CL (L/h), the dose (mg/m2) to be administered to a patient can be calculated using the product of CL (L/h/m2) and AUC. In order to reach AUC18 years, the dose applied to an 18-year-old (Dose18 years) is multiplied by the ratio of CLi to CL18 years (Eqs. 3, 4 and 5).
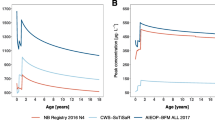
Therefore, the percentage of the protocol dose to be applied to children of different ages, given using the ratio of CLi and CL18 years, is displayed in Table 3. The CL (L/h/m2) of a 1-year-old child is predicted to be 67 % of that of an adult, and the dose (mg/m2) resulting in a comparable AUC would be 67 % of the full dose (mg/m2) administered to an 18-year-old (Table 3). CL was associated with an interindividual variability of approximately 31 % in the developed model. Thus, using this dosing proposal, AUCs in patients would be subject to the same degree of variability. For the impact of such a dose adjustment on the plasma concentration–time curve in a 1-year-old administered 20 mg/m2 over 4 h (see Fig. 2). While AUC is significantly lower, C max is also reduced by approximately 30 % in this example.

Plasma concentration–time curve of a 1-year-old child with median height and body weight with (blue line) and without (red line) the application of the developed dose reduction scheme; infusion duration 4 h, full BSA dose 20 mg/m2. BSA body surface area
The model suggests that due to the close agreement between CL18 years and CLi (86 vs. 100 %), dose adaptation would not be necessary over the age of 7 years (Table 2). Under the described proposal, a median AUC of 0.675 mg/L·h (range 0.52–2.24) could be achieved in children younger than 3 years of age, compared with 0.678 mg/L·h (range 0.41–1.52) in older children, when taking the EPOC population as a hypothetical example. Figure 1f illustrates that the dose adaptation based on the pharmacokinetic model could provide a smooth transition based on age.
When comparing the evaluated dose adaptations with the proposed dosing regimen, body-weight-based dose reductions perform slightly better than proportional reductions based on the BSA dose. The closest agreement was observed with protocol E (SIOPEN HR-NBL-1; Fig. 1e), which reduces the dose based on body weight in children up to 12 kg. However, our approach would propose dose reduction based on age instead of body weight, and recommend this be applied in children up to the age of 7 years (Table 3).
Besides all these considerations, the data of the trial and the associated model show that dosing guidelines should reflect model variability. Ninety percent of the estimated CL values of the EPOC trial were within a twofold range; however, the population included outliers with considerably lower CL values. Considering this, even developing a more sophisticated dosing rule might not result in the achievement of a target AUC in all patients, and variability may be more pronounced in the very young.
Thus, translating dose adaptations based on pharmacokinetics into acceptable proposals for tumor-specific treatment groups fundamentally requires the identification and validation of target parameters. These parameters should be suitable as surrogates for clinical endpoints of interest and must take into account probabilistic aspects.
6 Conclusions
The considerations presented here offer the opportunity to discuss how to achieve a model-informed dose-reduction for doxorubicin based on pharmacological data, in contrast to the commonly used empirical dose adaptations. Visualizing the effects of commonly applied protocols highlights the need for a consensus in dose recommendations in the very young as considerable differences among various protocols are apparent. It would be highly desirable to establish one consistent dosing strategy for all cancer entities, and to prospectively validate this strategy with regard to efficacy and safety in clinical trials.
The clinical setting of rare diseases, with very small patient numbers, vulnerable infants, and a drug with complex pharmacokinetics, does not lend itself easily to the running of subsequent trials. Therefore, the next trial should be based on optimal modeling and simulation processes [29] as well as a well-established consensus on the goals (Table 4). If anyone would like to participate in such a process, they are welcome to contact the authors. Do we need age-specific dose calculations to adapt to age-dependent pharmacokinetic parameters, to reflect age-dependent vulnerability of target organs or to hit a smaller therapeutic gain?
Notes
For the purpose of this article, we are using the broad term drug ‘exposure’ to refer to dose (drug input into the body—along the lines of the FDA Guideline “Exposure-Response Relationships—Study Design, Data Analysis, and Regulatory Applications”, 2003) and dose ‘intensity’ to refer to the plasma concentrations experienced by an individual (quantified as AUC or average concentration).
References
Voller S, Boos J, Krischke M, Wurthwein G, Kontny NE, Boddy AV, et al. Age-dependent pharmacokinetics of doxorubicin in children with cancer. Clin Pharmacokinet. 2015;54(11):1139–49.
van Dalen EC, Raphael MF, Caron HN, Kremer LC. Treatment including anthracyclines versus treatment not including anthracyclines for childhood cancer. Cochrane Database Syst Rev. 2014;(9):CD006647.
Pritchard-Jones K, Bergeron C, de Camargo B, van den Heuvel-Eibrink MM, Acha T, Godzinski J, et al. Omission of doxorubicin from the treatment of stage II-III, intermediate-risk Wilms’ tumour (SIOP WT 2001): an open-label, non-inferiority, randomised controlled trial. Lancet. 2015;386(9999):1156–64.
Mulrooney DA, Yeazel MW, Kawashima T, Mertens AC, Mitby P, Stovall M, et al. Cardiac outcomes in a cohort of adult survivors of childhood and adolescent cancer: retrospective analysis of the Childhood Cancer Survivor Study cohort. BMJ. 2009;339:b4606.
Armstrong GT, Kawashima T, Leisenring W, Stratton K, Stovall M, Hudson MM, et al. Aging and risk of severe, disabling, life-threatening, and fatal events in the childhood cancer survivor study. J Clin Oncol. 2014;32(12):1218–27.
Kremer LC, van Dalen EC, Offringa M, Ottenkamp J, Voute PA. Anthracycline-induced clinical heart failure in a cohort of 607 children: long-term follow-up study. J Clin Oncol. 2001;19(1):191–6.
Leger K, Slone T, Lemler M, Leonard D, Cochran C, Bowman WP, et al. Subclinical cardiotoxicity in childhood cancer survivors exposed to very low dose anthracycline therapy. Pediatr Blood Cancer. 2015;62(1):123–7.
Lipshultz SE, Lipsitz SR, Mone SM, Goorin AM, Sallan SE, Sanders SP, et al. Female sex and drug dose as risk factors for late cardiotoxic effects of doxorubicin therapy for childhood cancer. N Engl J Med. 1995;332(26):1738–43.
Crawford JD, Terry ME, Rourke GM. Simplification of drug dosage calculation by application of the surface area principle. Pediatrics. 1950;5(5):783–90.
Lee JW, Aminkeng F, Bhavsar AP, Shaw K, Carleton BC, Hayden MR, et al. The emerging era of pharmacogenomics: current successes, future potential, and challenges. Clin Genet. 2014;86(1):21–8.
McLeod HL, Relling MV, Crom WR, Silverstein K, Groom S, Rodman JH, et al. Disposition of antineoplastic agents in the very young child. Br J Cancer Suppl. 1992;18:S23–9.
Palle J, Frost BM, Peterson C, Gustafsson G, Hellebostad M, Kanerva J, et al. Doxorubicin pharmacokinetics is correlated to the effect of induction therapy in children with acute myeloid leukemia. Anticancer Drugs. 2006;17(4):385–92.
Thompson PA, Rosner GL, Matthay KK, Moore TB, Bomgaars LR, Ellis KJ, et al. Impact of body composition on pharmacokinetics of doxorubicin in children: a Glaser Pediatric Research Network study. Cancer Chemother Pharmacol. 2009;64(2):243–51.
Frost BM, Eksborg S, Bjork O, Abrahamsson J, Behrendtz M, Castor A, et al. Pharmacokinetics of doxorubicin in children with acute lymphoblastic leukemia: multi-institutional collaborative study. Med Pediatr Oncol. 2002;38(5):329–37.
Pinkel D. The use of body surface area as a criterion of drug dosage in cancer chemotherapy. Cancer Res. 1958;18(7):853–6.
Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology: drug disposition, action, and therapy in infants and children. N Engl J Med. 2003;349(12):1157–67.
Rodvold KA, Rushing DA, Tewksbury DA. Doxorubicin clearance in the obese. J Clin Oncol. 1988;6(8):1321–7.
Centers for Disease Control and Prevention, National Center for Health Statistics. CDC growth charts: United States. 2010. Available at: http://www.cdc.gov/growthcharts/. Accessed 22 Aug 2016.
McCune JS, Gibbs JP, Slattery JT. Plasma concentration monitoring of busulfan: does it improve clinical outcome? Clin Pharmacokinet. 2000;39(2):155–65.
Evans WE, Relling MV, Rodman JH, Crom WR, Boyett JM, Pui CH. Conventional compared with individualized chemotherapy for childhood acute lymphoblastic leukemia. N Engl J Med. 1998;338(8):499–505.
Newell DR, Pearson AD, Balmanno K, Price L, Wyllie RA, Keir M, et al. Carboplatin pharmacokinetics in children: the development of a pediatric dosing formula. The United Kingdom Children’s Cancer Study Group. J Clin Oncol. 1993;11(12):2314–23.
Barry E, Alvarez JA, Scully RE, Miller TL, Lipshultz SE. Anthracycline-induced cardiotoxicity: course, pathophysiology, prevention and management. Expert Opin Pharmacother. 2007;8(8):1039–58.
van Dalen EC, van der Pal HJ, Kok WE, Caron HN, Kremer LC. Clinical heart failure in a cohort of children treated with anthracyclines: a long-term follow-up study. Eur J Cancer. 2006;42(18):3191–8.
Bielack SS, Erttmann R, Winkler K, Landbeck G. Doxorubicin: effect of different schedules on toxicity and anti-tumor efficacy. Eur J Cancer Clin Oncol. 1989;25(5):873–82.
Von Hoff DD, Layard MW, Basa P, Davis HL Jr, Von Hoff AL, Rozencweig M, et al. Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med. 1979;91(5):710–7.
Lipshultz SE, Giantris AL, Lipsitz SR, Kimball Dalton V, Asselin BL, Barr RD, et al. Doxorubicin administration by continuous infusion is not cardioprotective: the Dana-Farber 91-01 Acute Lymphoblastic Leukemia protocol. J Clin Oncol. 2002;20(6):1677–82.
Wurthwein G, Boos J. Low dose–high dose: what is the right dose? Pharmacokinetic modeling of etoposide. Cancer Chemother Pharmacol. 2002;49(4):303–8.
Godoy LY, Fukushige J, Igarashi H, Matsuzaki A, Ueda K. Anthracycline-induced cardiotoxicity in children with malignancies. Acta Paediatr Jpn. 1997;39(2):188–93.
van Hasselt JG, van Eijkelenburg NK, Beijnen JH, Schellens JH, Huitema AD. Optimizing drug development of anti-cancer drugs in children using modelling and simulation. Br J Clin Pharmacol. 2013;76(1):30–47.
Acknowledgments
The authors wish to thank all clinical investigators and clinical trial centers that participated in the EPOC-MS-001-Doxo trial. This trial and all accompanying research were funded by the European Community’s Seventh Framework Programme (FP7/2009-2013) under Grant Agreement Number 222910. Miriam Krischke and Gudrun Würthwein are supported by the German Federal Ministry of Research and Education (BMBF Grant 01KN1105).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
In addition to the public project funding mentioned in the Acknowledgments section, no external funding was used in the preparation of this manuscript.
Conflict of interest
Swantje Völler, Georg Hempel, Gudrun Würthwein, Alan V. Boddy, Miriam Krischke, Nicolas André, Maurizio D’Incalci, Gianni Bisogno and Joachim Boos declare that they have no conflicts of interest that may be relevant to the contents of this article.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Völler, S., Hempel, G., Würthwein, G. et al. Towards a Model-Based Dose Recommendation for Doxorubicin in Children. Clin Pharmacokinet 56, 215–223 (2017). https://doi.org/10.1007/s40262-016-0451-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-016-0451-y