
Abstract
Background and Objectives
Infliximab is a monoclonal anti-tumor necrosis factor-α (anti-TNFα) antibody that profoundly modified the treatment of Crohn’s disease (CD). The polymorphism of Fc fragment of IgG, low affinity IIIa, receptor (CD16a) [FCGR3A] influences the biological response to infliximab in patients with CD. Our aim was to study its influence on infliximab pharmacokinetics and risk of relapse after infliximab discontinuation.
Methods
In 111 CD patients in remission, infliximab was discontinued and its concentrations were measured for 30 months or until relapse. Infliximab pharmacokinetics were described using monocompartmental population modeling.
Results
The elimination rate of infliximab increased with C-reactive protein (CRP) [p = 0.00018] and was 16 % higher in FCGR3A-158V/V patients than in F carriers (p = 0.0028). Risk of relapse was higher in patients with baseline CRP ≥5 mg/L than in those with a lower value (p = 0.0000029). In addition, there was a first-order interaction between CRP and the FCGR3A genotype; in patients with high CRP, risk of relapse was higher for V/V patients than for F carriers (hazard ratio 4.80 and 2.84 for V/V and F carriers, respectively; p = 0.013).
Conclusion
Both increased inflammation and FCGR3A-158V/V genotype are associated with increased infliximab elimination and risk of relapse after infliximab discontinuation in patients with CD.
Similar content being viewed by others


FCGR3A-158V/F polymorphism was known to influence the variability of the effect and concentration-effect relationship of therapeutic antibodies, with homozygous V/V patients being more sensitive to treatment than F carriers. In this study, we found faster infliximab elimination in FCGR3A-158V/V patients with Crohn’s disease (CD) than in F carriers. |
FCGR3A-158V/F was also known to influence infliximab efficacy in patients with CD; infliximab treatment is more effective in V/V patients than in F-carrier patients. In this study we found that this polymorphism enhanced the risk of relapse due to inflammation compared with other genotypes when infliximab treatment was discontinued. |
Infliximab may therefore be more effective but more ‘drug consuming’ in V/V patients than in F-carrier patients with CD, leading V/V patients to a faster underexposure to infliximab and relapse than F-carrier patients. |
1 Introduction
Infliximab is a chimeric immunoglobulin G1 (IgG1) monoclonal antibody targeting tumour necrosis factor-α (TNFα). This biopharmaceutical has profoundly modified the treatment of inflammatory diseases, notably rheumatoid arthritis (RA), ankylosing spondylitis (AS), ulcerative colitis (UC) and Crohn’s disease (CD).
Infliximab pharmacokinetics show a large interindividual variability in patients with RA [1], AS [2, 3], CD [4–6], and UC [7]. In patients with chronic inflammatory bowel disease (IBD), infliximab pharmacokinetics were studied using population pharmacokinetic modeling in three studies [5–7]; however, they were based on scarce data and the terminal elimination phase could not be studied properly.
The pharmacokinetic variability of infliximab is relevant because the rate of clinical response increases with its concentrations in CD [4, 8, 9], as well as in patients with RA [1, 10–12]. In addition, response to infliximab may be influenced by Fc fragment of IgG, low affinity IIIa, receptor (CD16a) [FCGR3A]-158V/F polymorphism. The FCGR3A gene, which encodes FcγRIIIA, a receptor binding the Fc portion of IgG, is expressed on macrophages and natural killer (NK) cells. It presents a single nucleotide polymorphism which generates two FcγRIIIA allotypes, with either a valine (V) or phenylalanine (F) at position 158. In vitro, human IgG have a higher affinity for FcγRIIIA-158V than for FcγRIIIA-158F receptors [13]. In vivo, the influence of FCGR3A polymorphism was first described in patients with follicular non-Hodgkin lymphoma treated with rituximab monotherapy, Cartron et al. [14] showed that V/V patients had a higher objective response rate and longer progression-free survival than F carriers, a result that was confirmed by Weng et al. [15] in a larger number of patients. This better clinical response of FCGR3A-158V/V patients compared with F carriers was reported for two other anticancer therapeutic antibodies—trastuzumab in breast cancer [16] and cetuximab in colorectal cancer [17]. In kidney transplant patients, FCGR3A polymorphism influences the relationship between the dose of anti-lymphocyte globulins and lymphocyte depletion, the V allele being associated with a higher sensitivity to the therapeutic antibody than the F allele [18, 19]. These antibodies were shown to induce cytolysis at least partly by antibody-dependent cellular cytotoxicity (ADCC); after binding of the Fab portion of the antibody to its target-antigen, the Fc portion of the antibody can bind to FcγRIIIA expressed at the surface of effector cells, leading to lysis of the target cell. An in vitro model of ADCC suggested that this genotype influenced the concentration-effect of rituximab. In this study, the concentration leading to 50 % of maximal lysis induced by rituximab (EC50) was about fourfold lower for FCGR3A-158V/V than for F/F donors [20]. However, to date, the influence of FCGR3A polymorphism on the pharmacokinetics of therapeutic antibodies was never investigated.
In patients with CD treated with infliximab, a better biological response was observed in V/V patients than in F carriers, with C-reactive protein (CRP) levels during treatment being lower in V/V patients [21–23]. Some in vitro [24, 25] and in vivo [23] studies suggested that infliximab is able to kill mucosal mononuclear cells through ADCC. Therefore, the fact that the FCGR3A genotype influences inflammatory response could be explained by infliximab-mediated ADCC.
The present work used data from a previously published study [26], which was designed to identify the individual factors predicting the risk of relapse following infliximab discontinuation in patients with CD. Based on multivariate lifespan analysis, risk factors for relapse were high levels of leukocyte counts, previous surgical resection, low hemoglobin level, high CRP level, fecal calprotectin, and male sex. However, in this study, neither the pharmacokinetics of infliximab nor the influence of FCGR3A polymorphism on infliximab pharmacokinetics or time to relapse was investigated. The present work was aimed at describing the elimination phase of infliximab in patients with CD, and at investigating the influence of FCGR3A polymorphism on infliximab pharmacokinetics and risk of relapse in patients with CD discontinuing their treatment.
2 Methods
2.1 Study Design
The present work is an ancillary study of a previously published study (NCT00571337) [26]. This prospective, multicenter cohort study, conducted in 20 centers in France and Belgium between March 2006 and December 2009, was designed to assess the risk of relapse in CD patients in remission following infliximab discontinuation. The study protocol and documents were approved by the Ethics Committee of the Saint-Louis Hospital in Paris on 26 May 2005. The Investigational Review Board at each of the participating centers approved the protocol. All patients provided written informed consent before screening, including for FCGR3A genotyping. Patients included in this study had (i) to be treated with infliximab and an antimetabolite agent (azathioprine, mercaptopurine, or methotrexate); (ii) to present a prospective Crohn’s Disease Activity Index (CDAI) ≤150; (iii) to have been stable for at least 3 months and had to have been in corticosteroid-free remission over the past 6 months before inclusion; and (iv) to have received at least 1 year of scheduled infliximab injections, and at least two infusions of infliximab had to have been administered during the past 6 months. Before relapse, follow-up visits were performed at 14 days and every 2 months after the last infliximab infusion up to 30 months, or until relapse, study withdrawal or closing date (31 December 2009). Blood samples and clinical data were collected at baseline and at each scheduled visit, or at the time of relapse. A total of 115 patients were analyzed in the previously included study. In the present study, we analyzed patients for whom FCGR3A genotyping was available, i.e. 111 patients.
2.2 Clinical Data
The CDAI was evaluated at each scheduled visit or at the time of relapse. A relapse was defined by a CDAI above 250 points or between 150 points and 250 points, with a 70-point increase from baseline over 2 consecutive weeks.
2.3 Biological Analyses
Hemoglobin, hematocrit, white blood cell (WBC) count, CRP and platelet count were measured by routine procedures. Blood samples were also collected just before the last infliximab infusion for central measurement of high-sensitivity CRP (hsCRP), infliximab trough concentrations and antibodies toward infliximab (ATI) detection. Genotyping of FCGR3A was performed as previously described [27]. ATI were analyzed using a double-antigen enzyme-linked immunosorbent assay based on their capture by infliximab-coated microplates and their detection by peroxidase-conjugated infliximab. Anti-infliximab antibodies were therefore inconclusive when the serum infliximab level was >1 mg/L, negative when the concentration was <0.07 mg/L and the serum infliximab level was <1 mg/L, and positive when the concentration of anti-infliximab antibodies was >0.07 mg/L.
2.4 Infliximab Concentrations
Concentrations of infliximab were measured using a validated enzyme-linked immunosorbent assay [28]. Briefly, recombinant human TNFα was coated on the solid phase and recognized by infliximab, and the therapeutic antibody was detected by an anti-human IgG Fcγ-specific antibody conjugated to horseradish peroxidase. The limit of detection was 0.014 mg/L, and the lower [LLOQ] and upper limits of quantification [ULOQ] (between-assay accuracy, CV %) were 0.04 mg/L (9.8 %) and 4.5 mg/L (5.3 %), respectively. Sera exceeding the ULOQ were diluted 1:10 or 1:100.
2.5 Missing Data
Baseline hsCRP values were missing in five patients and were replaced by corresponding baseline non-hypersensitive CRP measurements. Pre-inclusion infliximab trough concentrations were missing in six patients (two for whom blood samples were not available, and four who were sampled after infliximab infusion), and their values were replaced by model predictions. ATI were not tested as a covariate because their presence was confirmed in only two patients. Pre-inclusion fecal calprotectin levels and erythrocyte sedimentation rates (ESRs) were missing in 27 (24 %) and 40 (35 %) patients, respectively, and were not analyzed in this study.
2.6 Pharmacokinetic Analysis
2.6.1 Software
Pharmacokinetic data were analyzed by a population approach using the non-linear mixed-effects program MONOLIX 4.2.2 software, which combines the stochastic expectation-maximization (SAEM) algorithm and a Markov Chain Monte Carlo procedure for likelihood maximization [29]. To ensure the best possible convergence, 600 K1 and 200 K2 (where K1 and K2 refer to the SAEM procedure of MONOLIX, known as ‘iteration kernels’) and simulated annealing were used to impose the convergence of the SAEM algorithm toward the global maximum of the likelihood. Fisher information matrix was computed using stochastic approximation. The objective function, which is the −2Ln likelihood (−2LL), was computed using importance sampling.
2.6.2 Data Below the Quantitation Limit
Because 25.6 % of infliximab concentrations were below the quantitation limit (BQL) (0.04 mg/L), BQL concentrations were interval-censored as being superior to 0 and inferior to 0.04 mg/L. Indeed, censoring BQL data was shown to be the best strategy to provide unbiased parameter estimates in the presence of data containing BQL concentrations [30].
2.6.3 Structural Model Design
Infliximab concentrations were described using compartmental pharmacokinetic models. One and two mamilliary models with first-order distribution constants were tested. Linear and non-linear (Michaelis–Menten) eliminations were also tested. Structural models were compared using Akaike’s information criterion (AIC), defined as AIC = OFV + 2 × p, where OFV is the value of the objective function and p is the number of model parameters to estimate. The OFV was −2 × ln-likelihood (−2LL). The model with the lowest AIC was selected.
2.6.4 Interindividual Models
The interindividual variability of pharmacokinetic parameters was described using an exponential model: for each parameter, interindividual variance was removed if it could not be properly estimated.
2.6.5 Error Model
Additive, proportional and mixed additive–proportional models were tested. For example, the combined additive–proportional model was implemented as follows: Y O,ij = Y P,ij.(1 + ε prop,ij) + ε add,ij where Y O,ij and Y P,ij are observed and predicted jth measurements for the ith patient, respectively, and ε prop,ij and ε add,ij are proportional and additive errors, with mean 0 and variances σ prop 2 and σ add 2, respectively.
2.6.6 Covariates
Covariates for which more than 10 % of values were missing were not assessed in this study. The influence of a binary covariate (CAT) on θ TV was implemented as ln(θ TV) = ln(θ CAT=0) + β CAT=1, where θ CAT=0 is the value of θ for an arbitrary reference category, and β CAT=1 is the value of θ TV for the other category. Continuous covariates (COV) were centered on their median, as follows: θ i = θ 0 × (COV/med(COV))βcov, where θ 0 is the value of θ for a median subject, β COV quantifies the influence of COV on θ, and med(COV) is the median value of COV in the population.
2.6.7 Model Comparison and Covariate Selection
Interindividual, residual and covariate models were compared using OFV and AIC. From pairs of nested models, the one with the lowest OFV was selected. This was assessed by a likelihood ratio test (LRT) in which the difference in OFV between two models (ΔOFV) is assumed to follow a Chi-square distribution with one degree of freedom. The influence of patient characteristics (covariates) was assessed in two steps:
-
Univariate step: The influence of each factor on pharmacokinetic parameters associated with interindividual variability was tested. Covariates were separately included into the base model. Covariates showing a significant influence (p < 0.1) were included in the model (full model).
-
Multivariate step: A backward stepwise elimination was performed: the covariates of the full model were removed one by one. Covariates whose removal resulted in a statistically significant increase in the OFV (p < 0.01) were retained in the model.
2.6.8 Model Goodness-of-Fit and Evaluation
The goodness-of-fit was assessed for each model by plotting population-predicted (PRED) and individually predicted (IPRED) concentrations versus observed concentrations (DV) and IPRED and DV versus time, and by evaluating the residuals by graphical inspection of population (PWRES) and individual-weighted residual distributions (IWRES), and normalized prediction distribution errors (NPDEs) [31].
2.7 Time-to-Relapse Analysis
Survival analysis was performed considering confirmed relapses as events. Time to relapse was calculated as the delay between the day of infliximab last dose and the day of relapse after infliximab discontinuation. Data censoring has been described previously [26]. If a patient had not relapsed, time progression was censored at closing date. Follow-up was censored in patients with ignored suspicion of relapse, unconfirmed relapse, or two successive missed visits.
The hazard ratios (HRs) were estimated by multivariate Cox proportional hazards regression model. Base model was the multivariable Cox model described previously [26], where factors related to time to relapse were corticosteroid use between 12 and 6 months before baseline (CORT), previous surgical resection (RESEC), male sex (SX), baseline hemoglobin level (HB) ≤145 g/L, leukocyte count (LC) >6.109 L−1, and Crohn’s Disease Endoscopic Index of Severity (CDEIS) >0 [26].
In addition, the influence of baseline hsCRP ≥5 mg/L, infliximab trough concentration and FCGR3A-158V/V genotype, as well as first-order interaction between these three factors, were tested in the multivariate model. The influence of factors was tested using Wald’s test. Time-to-relapse curves were estimated using the Kaplan–Meier method. Analysis of time-to-relapse data was made using R 3.0.1 (R Foundation for Statistical Computing, Vienna, Austria).
3 Results
3.1 Patients
The present work used data from a previously published study (NCT00571337) [26]. In the present study, of the 111 eligible patients, 50 relapses were confirmed (Table 1). Because their doses and dosing schedule had remained stable for at least 6 months, these patients were assumed to be at steady state of their infliximab concentrations.
3.2 Pharmacokinetic Analysis
A total of 546 serum samples were available for pharmacokinetic analysis, 150 (25.6 %) being BQL. The best description of infliximab concentrations was obtained using a one-compartment model with volume of distribution (V d) and first-order elimination rate constant (k e). Because early attempts showed that V d was strongly correlated with body weight (R 2 > 90 %), V d was estimated in liters per kilogram of body weight, and interindividual variance for V d (ω 2 Vd) was fixed to 0. Parameters describing a second compartment or a non-linear elimination were not identifiable. The best error model was proportional. Plots of predicted versus observed concentrations showed that the model agreed well with the data (Fig. 1). The PWRES, IWRES and NPDE plots showed that there was no obvious model mis-specification (Fig. 1). The pharmacokinetic parameters were estimated with good accuracy (Table 2). Typical values for V d and k e were 0.087 L/kg and 0.049 day−1, respectively. For a median-weighted (67 kg) patient, V d was therefore 6.1 L.

Upper: observed versus population model-predicted (a), observed versus individual model-predicted (b), and visual predictive checks (c) for infliximab concentrations. Full circles are infliximab concentration, solid line is median prediction and gray surface is 5–95 % confidence interval. Lower: population (d) and individual (e) residuals vs. population and individual concentration estimations, respectively, and normalized prediction distribution errors vs. Gaussian (f). PRED population predicted concentrations, IPRED individually predicted concentrations, DV observed concentrations, IWRES individual weighted residuals, WRES weighted residuals
Since ω 2 Vd was fixed to 0, no covariate was tested on V d. The influence of the following covariates was tested on k e using the difference of objective function value (ΔOFV, see Materials and Methods section) between two models:
-
COV at inclusion: age, body weight (WT), hemoglobin levels, WBC counts, aspartate aminotransferase (AST) and alanine aminotransferase (ALT), pre-inclusion and hsCRP concentrations.
-
discrete covariates: sex (SX) and the FCGR3A genotype (V/V vs. F carriers).
The univariate step indicated that k e was influenced by sex, body weight, hsCRP and the FCGR3A genotype. In the final model, k e increased with hsCRP (ΔOFV = 13.6; p = 0.00018), and was higher in patients with the FCGR3A-158V/V genotype than in F carriers (ΔOFV = 8.9; p = 0.0028) [Table 2; Fig. 2]. Elimination half-lives (t ½) of V/V and F-carrier patients were 12.2 and 14.1 days, respectively. In typical F carriers with pre-inclusion hsCRP values of 2 and 10 mg/L, t ½ was 14.2 and 13.0 days, respectively.

Distributions of typical parameters (upper) and random effects (lower) vs. covariates hsCRP (left) and FCGR3A genotype (right). The boxes represent the 25th, 50th and 95th percentiles, and the whiskers represent the 5th and 95th percentiles. hsCRP high-sensitivity C-reactive protein
3.3 Time-to-Relapse Analysis
Median time to relapse was 16 months. Base model was the multivariable Cox model previously described [26]. In addition to this model, we tested the influence of infliximab trough concentrations and the FCGR3A genotype. Baseline infliximab trough concentration and FCGR3A-158V/F genotype were not associated with time to relapse. Baseline hsCRP was associated with time to relapse, with patients with hsCRP ≥5 mg/L having a higher risk of relapse than those with a lower value [HR associated with hsCRP ≥5 mg/L (HRCRP≥5) = 2.84 (1.84–4.41), p = 0.0000029 (Table 3)]. In addition, a significant first-order interaction between hsCRP ≥5 and the FCGR3A genotype was observed regarding time to relapse (p = 0.013): HRCRP≥5 was higher for V/V patients than for F carriers (HRCRP≥5 = 4.80 and 2.84 for V/V and F carriers, respectively; Table 3). For F carriers, median time to relapse was 6.8 and 26.7 months for CRP ≥5 and <5 mg/L, respectively, and for V/V patients, median time to relapse was 4.9 and was not reached (>33 months) for hsCRP ≥5 and <5 mg/L, respectively (Fig. 3).

Kaplan–Meier curves of time to relapse according to FCGR3A genotype (V/V vs. F carriers) and baseline hsCRP (hsCRP < vs. ≥5 mg/L). hsCRP high-sensitivity C-reactive protein
4 Discussion
This is the first study showing an influence of FCGR3A-158V/F polymorphism on the pharmacokinetics of a therapeutic antibody. In our study, infliximab concentrations were satisfactorily described by a one-compartment model, and pharmacokinetic parameters were reliably estimated. In addition, to our knowledge this is the first study where several infliximab concentrations were available to describe the terminal elimination phase of this monoclonal antibody. The observed increase of V d of infliximab with body weight is in agreement with previous studies in other conditions [2, 3, 6, 7].
We observed an increase in infliximab elimination rate with pretreatment CRP concentrations (Table 2; Fig. 2). The effect of CRP levels was primarily shown to influence infliximab concentrations [10, 12], with infliximab trough concentrations at steady-state being inversely correlated with pre-inclusion CRP in patients with RA. We then showed that, in patients with RA, CRP concentrations actually influenced infliximab elimination [19]. Such an influence was never reported in patients with CD. The relationship between pre-inclusion CRP levels and infliximab elimination rate constant may be explained by two mechanisms. First, it can be explained by target-mediated drug disposition (TMDD), a mechanism frequently reported for monoclonal antibodies [32, 33]. Production of TNFα leads to increased CRP concentration, therefore serum CRP may be considered as an indirect marker of TNFα concentration [34–36]. Consequently, in patients with substantial inflammation, the relationship between CRP and infliximab elimination rate constant may be explained by an increase in infliximab target-mediated elimination rate constant related to antigenic burden (TNFα levels; Fig. 4), with infliximab being ‘consumed’ by TNFα. It can also be explained by the infliximab concentration-effect relationship. Indeed, because the patients were at steady state, this influence could also be explained by the fact that higher exposure to infliximab led to lower TNFα and therefore CRP concentrations.

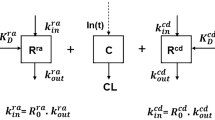
Putative target-mediated drug disposition of infliximab in Crohn’s disease. Upper: interaction between infliximab and serum TNF. Lower: interaction between infliximab and TNF expressed at the membrane of inflammatory cells. Because of the higher affinity of the V allotype of FcγRIIIA for the Fc portion of IgGs, the kADCC may be higher in V/V patients than in F carriers, resulting in an increased target-mediated infliximab elimination. IFX infliximab, TNF tumor necrosis factor-α, IFX-TNF infliximab-TNF complex, NK natural killer cells, k ADCC infliximab-induced antibody-dependent cell cytotoxicity elimination rate constant, IgG immunoglobulin G, k deg elimination rate constant of infliximab-circulating CRP complexes, k e elimination rate constant of unbound infliximab, In(t) infliximab infusion
In addition, we observed a higher infliximab elimination rate constant in FCGR3A-158V/V patients than in F carriers (0.057 and 0.049 day−1, respectively; Table 2). The FCGR3A polymorphism was first found to influence the clinical response to therapeutic antibodies acting, at least in part, by ADCC. The higher affinity of the FcγRIIIA V allele than the F allele for the Fc portion of IgGs results in higher NK cell recruitment in V/V patients than in F carriers [13]. This may explain (i) the higher potency of rituximab in inducing ADCC with NK cells from V/V than from F/F donors [20], and (ii) the better clinical response of patients with non-Hodgkin lymphoma treated with rituximab [14, 15] and cancer patients treated with other antibodies acting partly by ADCC [16, 17]. The influence of the FCGR3A genotype on infliximab pharmacokinetics may be explained by TMDD (Fig. 4). Indeed, in addition to targeting circulating TNFα, infliximab may opsonize TNFα-expressing cells and recruit cytotoxic (notably NK) cells, this recruitment leading to the lysis of target cells by ADCC [24, 25]. In TMDD models [32, 33], the elimination of drug-target complexes is usually described using a first-order k e (usually noted as k int, for ‘internalization’), which assumes an elimination rate proportional to the amount of antibody-target complex. Increasing k int would result in an increased antibody target-mediated elimination and therefore in antibody ‘total’ elimination. Assuming that target-cell ADCC may also be described using a first-order rate (k ADCC; Fig. 4), the higher infliximab elimination rate in V/V patients than in F-carrier patients may be explained by an increase in NK recruitment and therefore increased k ADCC in V/V patients. Thus, infliximab-induced ADCC may be more potent but also more ‘infliximab consuming’ in V/V patients than in F-carrier patients.
Even if non-linear elimination was never reported for infliximab, an increase in infliximab clearance (CL) for decreasing doses was reported [37], which suggests an influence of antigenic target, and therefore TMDD kinetics. However, since infliximab elimination was linear, TMDD models could not be assessed. Under the quasi-steady-state equilibrium hypothesis, the non-linear elimination term is written as: R T × k int × C/(K D + C), where C is infliximab concentration, R T is the total amount of antigen target, and K D is the dissociation rate constant. This term may be rewritten using a Michaelis–Menten term as follows: V m × C/(K m + C), with V m = R T × k int, where V m is the maximum elimination rate and K m is the Michaelis constant. For decreasing infliximab concentrations, the influence of this term on elimination increases. Using this model, both CRP and FCGR3A should influence V m and/or K m. However, V m and K m cannot be indentified for linear pharmacokinetics; the influence of CRP and FCGR3A may be deported on k e. However, the influence of these covariates on k e is an approximation of their influence on V m and/or K m because k e is a first-order parameter, unlike V m and K m.
In the analysis of time-to-relapse data, we confirmed that high (≥5 mg/L) baseline hsCRP levels were associated with an increased risk of relapse (HR 2.5). This may be related to increased infliximab consumption in patients with high inflammation; these patients may therefore be underexposed sooner than patients with low inflammation, leading to a shorter time to relapse.
In addition, we found a significant first-order interaction between FCGR3A and CRP levels, even if no significant influence of FCGR3A on risk of relapse was observed. Indeed, for patients with an hsCRP level ≥5 mg/L, the risk of relapse was higher for V/V patients than for F carriers (measured by the term of interaction, i.e. RRV/V–F = 1.7), whereas this difference was not significant for patients with an hsCRP level <5 mg/L. Therefore, the FCGR3A-158V/V genotype appears to enhance the risk of relapse due to inflammation compared with other genotypes. This appears paradoxical since, in CD, infliximab treatment is more effective [21–23] in V/V patients than in F-carrier patients. However, this finding is consistent with the influence of the FCGR3A genotype in infliximab pharmacokinetics. Patients with high inflammatory disease may present higher antigenic burden than other patients. This implies that infliximab treatment may be more effective but more ‘drug consuming’ in V/V patients than in F-carrier patients, leading V/V patients to a faster underexposure to infliximab and relapse than F-carrier patients. In other terms, V/V patients may respond better to infliximab treatment than F carriers, but infliximab withdrawal may lead to an increased risk of relapse in V/V, especially in patients with high inflammatory activity. In addition to underexposure to infliximab, this increased risk of relapse might be due to the turnover of TNFα-expressing cells, i.e. neutrophils, monocytes, lymphocytes [38–40]. The turnover of leukocytes, including neutrophils, was previously described using transit Friberg models, where precursor cell (first-order) proliferation is regulated due to mature cells, decreasing mature cell amount and resulting in an increase of precursor proliferation and vice versa [41–43]. Assuming this regulation pattern for TNFα-expressing cell regulation, an increased kADCC in V/V patients compared with F carriers may result in an increased precursor cell proliferation and therefore a shorter delay for TNFα-expressing cell recovery. However, this result should be confirmed in other studies and for other cytolytic therapeutic antibodies acting by ADCC.
Even if our study provided the first descriptions of the elimination phase of infliximab and the influence of the FCGR3A genotype on monoclonal antibody pharmacokinetics in a fair number of patients, it has several limitations.
-
First, the absence of blood samples between baseline and the 14th day after infliximab infusion. Indeed, 14 days after infliximab infusion, the distribution phase was over for all patients (Fig. 1). Therefore, the data may have provided insufficient information to identify the parameters of a peripheral pharmacokinetic compartment. Since infliximab pharmacokinetics were described using two-compartment models in most studies [2, 3, 5–7], our results are difficult to compare with those of other studies. However, the estimated t ½ of infliximab was 14.1 days, which is consistent with these studies. In addition, k e had to be used as a primary pharmacokinetic parameter instead of CL. Indeed, a typical estimate of CL was associated with rather important shrinkage (67 %) and a large correlation between V d and CL estimates (89 %). Of note, the elimination rate constant (k e) is not equal to the elimination rate constant (k 10), but equal to the elimination slope (β) which is estimated using a two-compartment model. Therefore, it is not possible to determine precisely which parameters (between elimination and transfer rate constants) are influenced by the FCGR3A genotype.
-
Second, a strong correlation (R = 0.96 %; data not shown) of body weight with V d led to an interindividual variance of V d that was not identifiable and therefore for which no covariate could be tested.
-
Third, the presence of ATI was previously reported to increase infliximab CL [2, 3, 5–7, 19]. In this study, the number of patients with ATI (two patients) was too low to analyze the effect of ATI on the infliximab elimination rate constant. Indeed, since ATI + patients are more likely to discontinue treatment [44], these patients may be underrepresented in the present study, in which only patients in long-term sustained remission were included. In addition, because ATI results were non-conclusive for infliximab concentrations >1 mg/L, false negative results may have occured, which may have participed to an underestimation of the porportion of immunized patients. Because the risk of developing ATI may be increased in patients with low concentrations of infliximab, which may have led to an underestimation of the number of patients with ATI and to a bias in the association of the FCGR3A genotype and hsCRP concentration with infliximab elimination. Indeed, hsCRP >5 mg/L and the V/V genotype were associated with increased infliximab elimination, while it was suggested that low infliximab concentrations increased the risk of developing ATI [44].
-
Fourth, unfortunately a full TMDD model (Fig. 4) could not be tested because neither circulating TNFα levels nor cell-expressing TNFα counts were available.
-
Finally, infliximab trough concentration before infusion was not associated with time to relapse. One explanation may be that patients from this study were treated for a mean of 2.2 years, and all were in remission; therefore, each patient may have had an infliximab trough concentration already adapted to his/her inflammatory activity.
5 Clinical Implications
This study showed for the first time that inflammation and FCGR3A polymorphism influence both infliximab pharmacokinetics and time to relapse in patients with CD; V/V patients treated with infliximab may respond better than F carriers, but infliximab withdrawal in these patients led to decreased infliximab concentrations and an increased risk of relapse, especially in the presence of high inflammatory activity. It might therefore be useful to account for FCGR3A polymorphism in the decision of infliximab withdrawal in these patients.
6 Conclusions
Our study is the first to show that FCGR3A polymorphism influences not only the efficacy but also the pharmacokinetics of infliximab. This influence could be due to an alteration of infliximab mediated by inflammatory cells expressing TNFα at their surface. This phenomenon may apply to other therapeutic antibodies acting partly by ADCC (e.g. rituximab, cetuximab, and trastuzumab). Nevertheless, this result needs to be confirmed for the other cytolytic antibodies and should be more precisely quantified using pharmacokinetic–pharmacodynamic studies where target-mediated CL of antibodies can be included in the model.
References
St Clair EW, Wagner CL, Fasanmade AA, Wang B, Schaible T, Kavanaugh A, et al. The relationship of serum infliximab concentrations to clinical improvement in rheumatoid arthritis: results from ATTRACT, a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002;46(6):1451–9.
Ternant D, Mulleman D, Lauferon F, Vignault C, Ducourau E, Wendling D, et al. Influence of methotrexate on infliximab pharmacokinetics and pharmacodynamics in ankylosing spondylitis. Br J Clin Pharmacol. 2011;2011(22):1365–2125.
Xu Z, Seitz K, Fasanmade A, Ford J, Williamson P, Xu W, et al. Population pharmacokinetics of infliximab in patients with ankylosing spondylitis. J Clin Pharmacol. 2008;48(6):681–95 (Epub 2008 Apr 9).
Baert F, Noman M, Vermeire S, Van Assche G, D’ Haens G, Carbonez A, et al. Influence of immunogenicity on the long-term efficacy of infliximab in Crohn’s disease. N Engl J Med. 2003;348(7):601–8.
Fasanmade AA, Adedokun OJ, Blank M, Zhou H, Davis HM. Pharmacokinetic properties of infliximab in children and adults with Crohn’s disease: a retrospective analysis of data from 2 phase III clinical trials. Clin Ther. 2011;33(7):946–64.
Ternant D, Aubourg A, Magdelaine-Beuzelin C, Degenne D, Watier H, Picon L, et al. Infliximab pharmacokinetics in inflammatory bowel disease patients. Ther Drug Monit. 2008;30(4):523–9.
Fasanmade AA, Adedokun OJ, Ford J, Hernandez D, Johanns J, Hu C, et al. Population pharmacokinetic analysis of infliximab in patients with ulcerative colitis. Eur J Clin Pharmacol. 2009;65(12):1211–28.
Maser EA, Villela R, Silverberg MS, Greenberg GR. Association of trough serum infliximab to clinical outcome after scheduled maintenance treatment for Crohn’s disease. Clin Gastroenterol Hepatol. 2006;4(10):1248–54.
Van Assche G, Magdelaine-Beuzelin C, D’Haens G, Baert F, Noman M, Vermeire S, et al. Withdrawal of immunosuppression in Crohn’s disease treated with scheduled infliximab maintenance: a randomized trial. Gastroenterology. 2008;134(7):1861–8.
Bendtzen K, Geborek P, Svenson M, Larsson L, Kapetanovic MC, Saxne T. Individualized monitoring of drug bioavailability and immunogenicity in rheumatoid arthritis patients treated with the tumor necrosis factor alpha inhibitor infliximab. Arthritis Rheum. 2006;54(12):3782–9.
Mulleman D, Chu Miow Lin D, Ducourau E, Emond P, Ternant D, Magdelaine-Beuzelin C, et al. Trough infliximab concentrations predict efficacy and sustained control of disease activity in rheumatoid arthritis. Ther Drug Monit. 2010;32(2):232–6.
Wolbink GJ, Voskuyl AE, Lems WF, de Groot E, Nurmohamed MT, Tak PP, et al. Relationship between serum trough infliximab levels, pretreatment C reactive protein levels, and clinical response to infliximab treatment in patients with rheumatoid arthritis. Ann Rheum Dis. 2005;64(5):704–7.
Koene HR, Kleijer M, Algra J, Roos D, von dem Borne AE, de Haas M. Fc gammaRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell Fc gammaRIIIa, independently of the Fc gammaRIIIa-48L/R/H phenotype. Blood. 1997;90(3):1109–14.
Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99(3):754–8.
Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 2003;21(21):3940–7.
Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol. 2008;26(11):1789–96.
Zhang W, Gordon M, Schultheis AM, Yang DY, Nagashima F, Azuma M, et al. FCGR2A and FCGR3A polymorphisms associated with clinical outcome of epidermal growth factor receptor expressing metastatic colorectal cancer patients treated with single-agent cetuximab. J Clin Oncol. 2007;25(24):3712–8.
Ternant D, Buchler M, Beneton M, Alvan G, Ohresser M, Touchard G, et al. Interindividual variability in the concentration-effect relationship of antilymphocyte globulins: a possible influence of FcgammaRIIIa genetic polymorphism. Br J Clin Pharmacol. 2008;65(1):60–8.
Ternant D, Ducourau E, Perdriger A, Corondan A, Le Goff B, Devauchelle-Pensec V, et al. Relationship between inflammation and infliximab pharmacokinetics in rheumatoid arthritis. Br J Clin Pharmacol. 2013;2013(19):12313.
Dall’Ozzo S, Tartas S, Paintaud G, Cartron G, Colombat P, Bardos P, et al. Rituximab-dependent cytotoxicity by natural killer cells: influence of FCGR3A polymorphism on the concentration-effect relationship. Cancer Res. 2004;64(13):4664–9.
Louis E, El Ghoul Z, Vermeire S, Dall’Ozzo S, Rutgeerts P, Paintaud G, et al. Association between polymorphism in IgG Fc receptor IIIa coding gene and biological response to infliximab in Crohn’s disease. Aliment Pharmacol Ther. 2004;19(5):511–9.
Louis EJ, Watier HE, Schreiber S, Hampe J, Taillard F, Olson A, et al. Polymorphism in IgG Fc receptor gene FCGR3A and response to infliximab in Crohn’s disease: a subanalysis of the ACCENT I study. Pharmacogenet Genomics. 2006;16(12):911–4.
Moroi R, Endo K, Kinouchi Y, Shiga H, Kakuta Y, Kuroha M, et al. FCGR3A-158 polymorphism influences the biological response to infliximab in Crohn’s disease through affecting the ADCC activity. Immunogenetics. 2013;65(4):265–71.
Scallon B, Cai A, Solowski N, Rosenberg A, Song XY, Shealy D, et al. Binding and functional comparisons of two types of tumor necrosis factor antagonists. J Pharmacol Exp Ther. 2002;301(2):418–26.
Scallon BJ, Trinh H, Nedelman M, Brennan FM, Feldmann M, Ghrayeb J. Functional comparisons of different tumour necrosis factor receptor/IgG fusion proteins. Cytokine. 1995;7(8):759–70.
Louis E, Mary JY, Vernier-Massouille G, Grimaud JC, Bouhnik Y, Laharie D, et al. Maintenance of remission among patients with Crohn’s disease on antimetabolite therapy after infliximab therapy is stopped. Gastroenterology. 2012;142(1):63–70.
Dall’Ozzo S, Andres C, Bardos P, Watier H, Thibault G. Rapid single-step FCGR3A genotyping based on SYBR Green I fluorescence in real-time multiplex allele-specific PCR. J Immunol Methods. 2003;277(1–2):185–92.
Ternant D, Mulleman D, Degenne D, Willot S, Guillaumin JM, Watier H, et al. An enzyme-linked immunosorbent assay for therapeutic drug monitoring of infliximab. Ther Drug Monit. 2006;28(2):169–74.
Monolix 4.2.2 user’s guide. Orsay: Lixoft SAS (2013). Available from: http://www.lixoft.eu/wp-content/resources/docs/UsersGuide.pdf. Accessed 27 Jul 2013.
Bergstrand M, Karlsson MO. Handling data below the limit of quantification in mixed effect models. AAPS J. 2009;11(2):371–80. doi:10.1208/s12248-009-9112-5.
Comets E, Brendel K, Mentre F. Computing normalised prediction distribution errors to evaluate nonlinear mixed-effect models: the npde add-on package for R. Comput Methods Programs Biomed. 2008;90(2):154–66.
Gibiansky L, Gibiansky E. Target-mediated drug disposition model: relationships with indirect response models and application to population PK-PD analysis. J Pharmacokinet Pharmacodyn. 2009;36(4):341–51. doi:10.1007/s10928-009-9125-9.
Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn. 2001;28(6):507–32.
Elliott MJ, Feldmann M, Maini RN. TNF alpha blockade in rheumatoid arthritis: rationale, clinical outcomes and mechanisms of action. Int J Immunopharmacol. 1995;17(2):141–5.
Elliott MJ, Maini RN. Anti-cytokine therapy in rheumatoid arthritis. Baillieres Clin Rheumatol. 1995;9(4):633–52.
Maini RN, Elliott MJ, Brennan FM, Williams RO, Chu CQ, Paleolog E, et al. Monoclonal anti-TNF alpha antibody as a probe of pathogenesis and therapy of rheumatoid disease. Immunol Rev. 1995;144:195–223.
FDA. Infliximab: clinical pharmacology review. Available from: http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/ucm107704.pdf. Accessed 21 Aug 2014.
Wright HL, Chikura B, Bucknall RC, Moots RJ, Edwards SW. Changes in expression of membrane TNF, NF-κB activation and neutrophil apoptosis during active and resolved inflammation. Ann Rheum Dis. 2011;70(3):537–43.
Horiuchi T, Mitoma H, Harashima S, Tsukamoto H, Shimoda T. Transmembrane TNF-alpha: structure, function and interaction with anti-TNF agents. Rheumatology. 2010;49(7):1215–28.
Mitoma H, Horiuchi T, Tsukamoto H, Tamimoto Y, Kimoto Y, Uchino A, et al. Mechanisms for cytotoxic effects of anti-tumor necrosis factor agents on transmembrane tumor necrosis factor alpha-expressing cells: comparison among infliximab, etanercept, and adalimumab. Arthritis Rheum. 2008;58(5):1248–57. doi:10.002/art.23447.
Friberg LE, Henningsson A, Maas H, Nguyen L, Karlsson MO. Model of chemotherapy-induced myelosuppression with parameter consistency across drugs. J Clin Oncol. 2002;20(24):4713–21.
Quartino AL, Friberg LE, Karlsson MO. A simultaneous analysis of the time-course of leukocytes and neutrophils following docetaxel administration using a semi-mechanistic myelosuppression model. Invest New Drugs. 2012;30(2):833–45.
Quartino AL, Karlsson MO, Lindman H, Friberg LE. Characterization of endogenous G-CSF and the inverse correlation to chemotherapy-induced neutropenia in patients with breast cancer using population modeling. Pharm Res. 2014;31(12):3390–403.
Ducourau E, Mulleman D, Paintaud G, Chu Miow Lin D, Lauféron F, Ternant D, et al. Antibodies toward infliximab are associated with low infliximab concentration at treatment initiation and poor infliximab maintenance in rheumatic diseases. Arthritis Res Ther. 2011;13(3):R105.
Acknowledgments
This work was a collaborative venture by GETAID (Groupe d’Étude Thérapeutique des Affections Inflammatoires du Tube Digestif). The following GETAID investigators participated in patient recruitment: Amiens (J.L. Dupas), Bordeaux (D. Laharie), Caen (J.M. Reimund), Clichy-Beaujon (Y. Bouhnik), Colombes – L Mourier (P. Jouet), Gent (M. De Vos), Liège (J. Belaiche, E. Louis), Lille (J.-F. Colombel, G. Vernier-Massouille), Lyon (S. Nancey), Marseille (J.C. Grimaud), Montpellier (M. Veyrac), Nantes (A. Boureille, M. Flamant), Paris – Hôpital Européen Georges Pompidou (R. Jian), Paris – Lariboisière (P. Marteau), Paris – St Louis (M. Lemann, M. Allez), Rouen (G. Savoye), Strasbourg (B. Duclos), and Tours (L. Picon). The authors would like to thank Jean-Yves Mary and Nicolas Azzopardi for statistical advice, Hervé Watier for scientific advice, Céline Desvignes for blood-sample management, Anne-Claire Duveau, Caroline Brochon, Audrey Farnault and Marie-Noëlle Marson for technical assistance with infliximab and ATI assays, and FCGR3A genotyping. The University Hospital of Tours received an FEDER (Fonds Européen de Développement Régional – European funding for regional development) for its CePiBAc (Cetre pilote de suivi biologique des anticorps thérapeutiques – Pilot centre for therapeutic antibodies monitoring). This work was partly supported by the French Higher Education and Research Ministry under the program ‘Investissements d’avenir’ Grant Agreement: LabEx MAbImprove ANR-10-LABX-53-01.
Disclosures
Jean-Frédéric Colombel has served as a consultant, advisory board member, or speaker for Abbvie, Amgen, Bristol Meyers Squibb, Celltrion, Ferring, Genentech, Giuliani SPA, Given Imaging, Janssen, Merck & Co., Millenium Pharmaceuticals Inc., Nutrition Science Partners Ltd, Pfizer Inc., Prometheus Laboratories, Receptos, Sanofi, Schering Plough Corporation, Takeda, Teva Pharmaceuticals, UCB Pharma, Vertex, and Dr. August Wolff GmbH & Co. Matthieu Allez has served as a consultant for Abbott, UCB Pharma, Ferring, Merck Sharp and Dohme Corp., TcLand, TxCell, Novo Nordisk, Pfizer, and GlaxoSmithKline; has received lecture fees from Merck Sharp and Dohme Corp., Ferring, Neovacs, Janssen Cilag, and Abbott; and has received financial support for research from Novo Nordisk. Edouard Louis has received fees for the following: research grant from AstraZeneca, Schering-Plough, and Abbott; speaker fees from Abbott, Abbvie, AstraZeneca, Ferring, Schering-Plough, MSD, Chiesi, Menarini, Nycomed, Falk, UCB; advisory board fees from Abbott, Abbvie, Ferring, UCB, MSD, Millenium, Mitsubishi Pharma, Takeda; and consultant fees from Abbvie. Gilles Paintaud has been a consultant for Laboratoires Français du Fractionnement et des Biotechnologies (LFB) and Pierre-Fabre Laboratories; his research team has received finance from Roche Pharma, Chugai, Pfizer, Novartis and Janssen. David Ternant, Zahir Berkane, Laurence Picon, and Valérie Gouilleux-Gruart declare no conflicts of interest.
Authors’ responsibility
David Ternant, the corresponding author, confirms full access to all data in the study, and final responsibility.
Authors’ contributions
David Ternant, Zahir Berkane, Laurence Picon, Valérie Gouilleux-Gruart, Jean-Frédéric Colombel, Matthieu Allez, Edouard Louis, and Gilles Paintaud wrote the manuscript; Edouard Louis and Gilles Paintaud designed the research; David Ternant, Zahir Berkane, and Gilles Paintaud analyzed the data; and Valérie Gouilleux-Gruart performed the analytical part.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ternant, D., Berkane, Z., Picon, L. et al. Assessment of the Influence of Inflammation and FCGR3A Genotype on Infliximab Pharmacokinetics and Time to Relapse in Patients with Crohn’s Disease. Clin Pharmacokinet 54, 551–562 (2015). https://doi.org/10.1007/s40262-014-0225-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-014-0225-3