
Abstract
Background and Objective
Despite its important role in cancer treatment, there is currently very limited available information concerning the clinical pharmacology of actinomycin D (Act D). The study was designed to characterise Act D pharmacokinetics and investigate the impact of pharmacogenetic variation on Act D disposition in children with cancer.
Methods
A total of 650 plasma samples collected over an 8 year period from 117 patients ≤21 years receiving Act D (0.4–1.6 mg/m2) were used to characterise a population pharmacokinetic model. Polymorphisms in ABCB1 were analysed in 140 patients.
Results
A 3-compartment model provided a good fit to the data. Median values for Act D clearance and volume of distribution in the central compartment (V 1) obtained from the model were 5.3 L/h and 1.9 L (13.9 L/h/70 kg and 7.5 L/70 kg), respectively. There was substantial inter-subject variation in all pharmacokinetic parameters (coefficients of variation 53–81 % for non-normalised values). Body weight was a major determinant of Act D clearance, such that dose capping at 2 mg in larger children at a protocol dose of 1.5 mg/m2 resulted in significantly lower area under the plasma concentration-time curves (mean AUC values: 9.3 versus 12.8 mg·min/L; P < 0.0001). No significant relationships were found between ABCB1 genetic variants and Act D pharmacokinetic parameters, nor between CL, V 1 or dose and incidence of grade 3 or 4 toxicity.
Conclusion
We have defined the pharmacokinetics of Act D in a paediatric patient population, providing robust estimates of key pharmacokinetic parameters. Pharmacokinetic data bring into question the current clinical practice of dose capping at 2 mg in larger patients. Pharmacogenetic variation in candidate drug transporter genes identified from preclinical studies does not significantly impact on Act D exposure in a clinical setting.
Similar content being viewed by others

We have characterised for the first time the pharmacokinetics of actinomycin D in a large patient population, providing robust estimates of pharmacokinetic parameters and identifying patient body weight as the major determinant of actinomycin D clearance in children with cancer. |
Pharmacokinetic data bring into question the clinical utility of dose capping at 2 mg in larger children, with significantly lower actinomycin D AUC values observed in these patients. This issue should be further investigated, particularly in relation to inconsistencies in dose capping levels across clinical trials and protocols, with actinomycin D capped at a higher dose of 2.5 mg in the US. |
Pharmacogenetic variation in ABCB1, a candidate drug transporter gene identified from in vitro and animal studies, does not significantly impact on actinomycin D pharmacokinetics in children. |
1 Introduction
Actinomycin D (Act D) is an anti-tumour antibiotic commonly used in the treatment of cancer in children and adults. As part of a multimodal approach, Act D is a key component in the treatment of Wilms tumour, where cure rates as high as 90 % have been achieved, rhabdomyosarcoma and Ewings sarcoma [1]. Act D binds to DNA in a guanine-dependent manner [2, 3], preventing the synthesis of RNA, and has been shown to inhibit the incorporation of nucleotide triphosphates into DNA [4].
Although treatment with Act D is often successful, one of the main challenges associated with Act D therapy is treatment-related hepatic toxicity or veno-occlusive disease, which occurs in up to 13.5 % of cases [5–7]. Age at treatment is a major risk factor, with patients less than 3 years old almost fourfold more likely to have some form of toxicity following Act D treatment as compared to children 3–21 years of age [8]. Recently, a retrospective study of over 3,000 patients confirmed the higher incidence of Act D-related toxicity in patients <1 year old. Toxicity was more likely to occur early in treatment, although the incidence was lower in patients who tolerated their first course of therapy [9]. Other major risk factors appear to be the presence of Wilms tumour, as opposed to other tumour types, and Act D dose intensity [5, 10]. Bearing these factors in mind, it is a concern that for many well established drugs such as Act D, currently used dosing guidelines are founded largely on empirical experience, as opposed to being based on a sound knowledge of the clinical pharmacology of the drug. Inconsistencies also exist between clinical protocols and tumour types. For example, dose capping of Act D at a maximum dose of 2 mg is generally employed in UK protocols, depending on tumour type, as compared to a dose cap of 2.5 mg in US protocols [9].
To date only one substantive paediatric patient population has been studied, with the data obtained used in multiple publications focusing on alternative pharmacokinetic modelling approaches [11, 23, 25]. The original study of 31 patients identified a large-degree of inter-patient variation in Act D plasma concentrations and exposure, with a greater than fourfold range in area under the plasma concentration-time curve (AUC0–6h) observed across the patient population. Smaller children generally had higher Act D AUC values and, despite the limited number of patients, this was linked to an increased risk of toxicity. However, this initial report was limited by low patient numbers and only sparse pharmacokinetic sampling out to 24 h after drug administration. Although more recent publications have reported population pharmacokinetic models based on data from 33 and 36 children, respectively [23, 25], in each case 31 of these patients were from the original pharmacokinetic study carried out in the UK [11]. Due to the limited data available therefore, the determination of reliable and robust pharmacokinetic parameters has clearly been challenging. Further characterisation of Act D pharmacokinetics, including an influence of pharmacogenetics, requires a significantly larger patient population and more intensive sampling.
ATP-binding cassette (ABC) transporters such as ABCB1, ABCC2 and ABCG2 are present at the apical membrane in the liver, kidney and intestine where they facilitate the removal of both endogenous and exogenous substrates via the bile, urine and faeces [12]. These proteins are also found at many important “sanctuary sites” such as blood–brain, blood–testis, and blood–placenta barriers [13–15]. In contrast, ABCC1 is present at the basolateral membrane of hepatocytes and proximal tubular cells, facilitating the export of compounds to the blood [16]. Many commonly-used anticancer drugs are substrates for ABC transporters, which have the potential to impact drug disposition and elimination from the body [17–21]. The influence of drug transporters on elimination from the body may be particularly relevant for a drug such as Act D, which does not appear to undergo significant metabolism [11, 22, 23].
Previously, we have demonstrated that cell lines over-expressing ABCB1, ABCC1 or ABCC2 exhibit decreased sensitivity to Act D as compared to the parental cell line. Of particular note, approximately 60-fold higher concentration for 50 % inhibition of cell proliferation values were observed in MDCKII cells overexpressing ABCB1, corresponding to significantly lower intracellular Act D concentrations. In line with these in vitro data, in vivo knockout of Abcb1a/1b resulted in higher Act D plasma and brain concentrations compared to those in wild-type mice [24]. In contrast to many other established anticancer drugs, no studies have currently been performed to investigate the impact of pharmacogenetic variation in ABCB1 on the pharmacokinetics of Act D in cancer patients.
In two large pharmacological trials conducted in the UK, patients less than 21 years old, receiving Act D as part of their standard treatment regimen were recruited. The major aim of these studies was to explore the potential influence of ABCB1 pharmacogenetic variation alongside a more definitive characterisation of the pharmacokinetics of Act D in children with cancer.
2 Patients and Methods
2.1 Study Population and Treatment
Study protocols were approved by the UK Trent Multicentre Research Ethics Committee and written informed consent was obtained from all patients or parents as appropriate. Eligible patients were under 21 years of age and were receiving Act D as part of standard chemotherapy for a range of tumour types. The studies were registered through the appropriate clinical trials registries (PK 2003 08-REC: 03/04/074, CTA: 23198/0001/001; PK 2006 07-REC 05/MRE04/62, CTA: 2005-002996-34, ClinicalTrials.gov identifier: NCT00900354) before opening to patient recruitment. Baseline toxicity data prior to Act D treatment, including baseline haemoglobin (Hb), white blood cell (WBC) and platelet counts, were obtained from patients’ notes and details of concomitant medications prior to and/or in combination with Act D were recorded. Additional patient characteristics and clinical parameters including glomerular filtration rate (GFR), creatinine, ALT and bilirubin measurements were also recorded following patient registration, i.e. prior to Act D pharmacokinetic sampling, for post-study analysis. The type of catheter used for Act D administration and pharmacokinetic sampling was also recorded for all patients studied, in part to address concerns relating to the previously reported issue of line ‘contamination’ following Act D sampling [25].
Act D was administered as a short intravenous infusion (1–5 min) at doses of 0.4–1.6 mg/m2, with the maximum dose capped at 2 mg for larger children. The dose of Act D administered was adjusted for infants aged <1 year, or weighing <10 kg in body weight, with protocol doses of 0.02–0.05 mg/kg. Toxicity following Act D treatment was assessed by the National Cancer Institute Common Terminology Criteria of Adverse Events (CTCAE v3) and recorded for all patients over a period of three weeks following Act D treatment.
2.2 Blood Sampling and Analysis
Blood samples (2 mL) for measurement of Act D concentrations were collected in heparinised tubes from a central venous line, prior to administration of Act D and at 5, 15 and 30 min, and 2, 4, 8, 24 and 26 h post administration. Prior to sampling, the central venous line was flushed according to a standardised procedure, to ensure negligible contamination of the sample from the administration fluid. This procedure included flushing the line with 10 mL saline immediately following Act D administration, with an additional flush with 5 mL saline prior to collection of the first Act D pharmacokinetic sample. Deadspace volumes were also taken and discarded prior to the collection of all samples for pharmacokinetic analysis. Actual sampling times were recorded along with details of the central line type used for sampling. Not all samples were available for all patients and more limited pharmacokinetic sampling was frequently carried out for smaller children. Blood samples were immediately centrifuged at 1,200g for 10 min at 4 °C. Plasma was separated and stored at −20 °C prior to analysis using a modified liquid chromatography–mass spectrometry (LC–MS) assay, with a limit of quantification of 0.25 ng/mL, as previously validated and described [11, 22]. Briefly, extraction of clinical samples was carried out with acetonitrile and analysis performed on an API 4000 LC/MS/MS (AB SCIEX) using an internal standard of 7-aminoactinomycin D. The method has been demonstrated to exhibit good reproducibility over a calibration curve range of 0.25–100 ng/mL, with intra- and inter-assay precision CVs of 2.7–11.3 and 2.3–7.8 %, respectively. Accuracy data from assay validation studies showed relative errors of 2.0–16.4 (intra-assay) and 10.4–15.2 % (inter-assay) [22].
2.3 Pharmacogenetic Analysis
Genomic DNA was obtained from whole blood samples using QIAamp DNA blood Maxi kits (Qiagen) according to the manufacturer’s instructions. DNA purity and concentration were measured using a NanoDrop ND-1000 (Thermo Scientific, Rockford, USA) and stored at −20 °C prior to pharmacogenetic analysis. Genotyping for ABCB1 SNPs 1236C>T (rs1128503), 3435C>T (rs1045642) and 2677G>T/A (rs2032582) was performed using TaqMan® probes and an ABI 7500 Fast Real-Time PCR System (Applied Biosystems, CA, USA) according to manufacturer’s instructions. Allelic discrimination was performed using Sequence Detection Software (Applied Biosystems, CA, USA).
2.4 Pharmacokinetic Analysis
Using NONMEM version 7.2 [26], a population pharmacokinetic model was fitted to Act D plasma concentration versus time data from a total of 650 samples obtained from 117 patients. Parameterisation of the final three compartment model was in terms of systemic clearance (CL) and central volume (V 1), two inter-compartmental clearances (Q 2 and Q 3) and two peripheral volumes (V 2 and V 3); the ADVAN11 and TRANS3 routines were used within NONMEM. The first order conditional estimation (FOCE) method with η–ε interaction was used to obtain parameter estimates and bootstrap confidence intervals were also obtained for the final model. Posthoc empirical Bayes estimates of CL and V 1 were obtained for each individual. A VPC was carried out for the final model for which 2,000 datasets were simulated. Time following drug administration was split into 12 bins. Bootstrapping and VPC were carried out using Perl Speaks NONMEM version 3.6.2 [27].
All population pharmacokinetic parameters were allometrically scaled [28]. The exponents were fixed to 0.75 and 1.0, for clearances and volumes respectively. Parameters were also scaled to a standard body weight of 70 kg so the model for the population parameters was:
where \( \theta_{i} \) is the typical value of the pharmacokinetic parameter (CL, Q 2, Q 3, V 1, V 2 or V 3) for an individual patient, \( \theta_{A} \) is the allometric exponent, WT i is patient weight and so \( \theta \) is the typical value for a hypothetical 70 kg patient. Random effects were added to pharmacokinetic parameters as appropriate, along with covariance parameters between these random effects. The model for those population parameters was as follows:
where \( \eta_{i} \) is the random effect for an individual patient. A composite intra-subject error model was used initially, but once a good model fit was obtained the additive component was found to be unnecessary and so in the final model a proportional intra-subject error model was utilised. Goodness of fit of the models was determined through examination of the NONMEM OFV, plots of individual predictions and observed plasma concentrations against time and examination of residuals.
2.5 Statistical Analysis
The association of covariates with CL and V 1 was assessed outside NONMEM using Stata/SE (StataCorp. 2009. Stata Statistical Software: Release 11.2. College Station, TX: StataCorp LP) as all of the covariates considered (creatinine, C1236T, G2677T/A, C3435T, toxicity grade, line type), with the exception of age and sex, contained missing values. While methods for handling missing data in non-linear mixed effects models are available, such as multiple imputation methods, the additional complexity entailed was not appropriate for this analysis. Thus, unexplained variation in CL and V 1 following allometric scaling was investigated by modelling η CLi (etaCL) and η V1i (etaV1), reflecting the difference between the estimated individual pharmacokinetic parameters and the estimated population values (adjusted for the known association between CL, V 1 and body weight). The distribution of etaCL and etaV1 was approximately Gaussian and so linear regression was used to assess the influence of continuous covariates and t-tests or ANOVA were used for categorical covariates. Genotype frequencies were assessed for Hardy–Weinberg equilibrium using the Pearson Chi-squared test. Differences in AUC values between patients receiving a dose of 1.5 mg/m2 as a capped versus non-capped dose were analysed by t-test following log transformation of data.
3 Results
3.1 Patient Characteristics
One hundred and fifty-eight patients receiving Act D as part of their standard chemotherapy regimen for various tumour types were recruited on to the studies between August 2004 and September 2012. The study population had a median age of 4.6 years (range 0.3–19.8 years) and included 78 male and 80 female patients. Wilms tumour was the most common tumour type (48 %), followed by rhabdomyosarcoma (30 %) and Ewing’s sarcoma (18 %). Other tumour types included a paraspinal undifferentiated sarcoma, an embryonal sarcoma of the liver, a pleuropulmonary blastoma, a metastatic primitive neuroectodermal tumour and a non-rhabdomyosarcoma tumour. Full patient characteristics can be found in Table 1. No bias was observed in terms of these patient characteristics when patients were divided into groups according to availability of pharmacokinetic data, pharmacogenetic data or both as shown in Table 1.
3.2 Actinomycin D Pharmacokinetics
Depending on treatment protocol, doses of Act D ranged from 0.19 to 2.5 mg (0.4–1.6 mg/m2). Out of the 158 patients recruited, blood samples for pharmacokinetic analysis were taken from 147 patients. Quality control data from our laboratory indicate that Act D is unstable in human plasma when stored at −20 °C for longer than six months and as such pharmacokinetic data from 14 patients were deemed invalid due to analysis taking place beyond this time period. Data from a further 14 patients were excluded due to artificially high Act D plasma concentrations, several magnitudes above the range observed at early time points following drug administration, indicative of sample contamination. In addition, data from two patients were excluded due to invalid assays and a single sample from an additional patient was excluded from analysis as the plasma concentration was below the LC–MS assay limit of quantitation of 0.25 ng/mL.
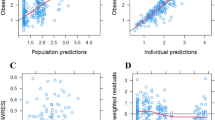
A total of 650 plasma samples from 117 patients had a valid analysis, and have been used to characterise the pharmacokinetic model. Figure 1a shows individual Act D plasma concentration (logarithmic scale) versus time data for all patients included in the model, with a summary of the pharmacokinetic data provided in supplementary Table S1. This table indicates the number of samples obtained at each study time point. Full sample sets with both early (5–30 min) and late (20–26 h) time points were provided by 82 patients (70 %).

Actinomycin D plasma concentration versus time data for all patients (n = 117) used in the population pharmacokinetic analysis (a) and individual weighted residuals (IWRES) versus time for population pharmacokinetic model 2 (b). The smoothed curves represented by dashed lines are generated using LOWESS (locally weighted scatterplot smoother)
A large degree of variability in plasma concentrations was observed among the 117 patients studied. For those patients sampled at 5 min the median concentration was 128 μg/L (range 64.7–186 μg/L, n = 13) and for those whose first sampling time was 15 min post administration the median was 24.4 μg/L (5.3–122 μg/L, n = 101). The median Act D plasma concentration in samples taken 24 h post-administration was 1.8 μg/L (0.7–4.8 μg/L, n = 73).
3.3 Population Pharmacokinetics
A population pharmacokinetic analysis using data from 117 patients was performed using NONMEM. Act D pharmacokinetics were characterised by a very short, rapid elimination phase, followed by an intermediate disposition phase and finally a slow terminal decline in plasma concentration, indicative of a three-compartment pharmacokinetic model. Any attempt to fit a two compartment model resulted in a poor fit and so the focus moved to three-compartment models. Initially the model included random effects on CL and V 1 together with a covariance parameter. Whilst this provided a reasonable fit, because of the overriding correlation between pharmacokinetic parameters and body weight, an allometrically-scaled model was developed with fixed exponents (Model 1). This model resulted in objective function value (OFV) drops of 773 from the comparable two-compartment model and 227 from the non-allometrically scaled three-compartment model. Even though it provided a much-improved fit, examination of residuals and individual fits demonstrated a systematic lack of fit around the 4 and 6 h time points where the model generally under-predicted Act D plasma concentrations. Including random effects for Q 3 and V 3 resulted in a drop in OFV of 62 and 96, respectively and allowing a full block covariance structure between random effects (excluding the correlation between CL and V 1) further improved the model.
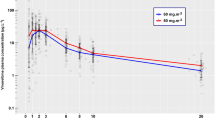
The final model (Model 2) thus contained random effects and correlation parameters for CL, Q 3 and V 3, and a further random effect for V 1. This final model provided a much-improved fit to the 4 and 6 h time points as seen in Fig. 1b. A comparison of model parameters is shown in supplementary Table S2, with bootstrap confidence intervals for the full model shown in Table 2. Visual Predictive Checks (VPCs) of the final model indicated that the model provided a reliable description of the observed data (Fig. 2). Median values of the empirical Bayes estimates of individual pharmacokinetic parameters were: clearance (CL) 5.3 L/h, inter-compartmental clearance (Q 2) 5.5 L/h, inter-compartmental clearance (Q 3) 13.9 L/h, central volume of distribution (V 1) 1.9 L, volume of distribution of the second compartment (V 2) 4.4 L and volume of distribution of the third compartment (V 3) 108 L. All pharmacokinetic parameters had large coefficients of variation (between 53 and 81 %) indicating substantial variation between patients. However, the unexplained variation after modelling was reduced to 28 % for both CL and V 1.

Visual predictive check for final model showing observed Actinomycin D plasma concentrations. The solid line shows the 50th percentile of the simulated data and the dashed lines show the 2.5th and 97.5th percentiles. The visual predictive check plots obtained after stratification by weight demonstrated a similar level of concordance between observed and simulated data
Creatinine measurements taken immediately prior to Act D treatment were available for 113 of the 117 patients used in the pharmacokinetic analysis, with a median creatinine value of 37 μmol/L (range 12–90 μmol/L). No association of serum creatinine was found with either etaCL or etaV1. The type of catheter used to administer Act D was investigated to address concerns relating to previously reported line ‘contamination’ issues [25]. Although catheter type appeared to have a minor influence on both CL and V 1, this was due to the association of line type with patient age and thus body size. Once patient body weight had been appropriately accounted for, the effect of line type was no longer observed (Fig. 3).

Lack of effect of central venous catheter line-type on the relationship between patient body weight and Actinomycin D clearance
Analysis of Act D AUC values for all patients who received a dose of 1.5 mg/m2 showed a significantly lower mean AUC value for larger children (>1.3 m2), where doses were capped at 2 mg, as compared to smaller children (≤1.3 m2) where dose capping was not required (mean AUC values: 9.3 versus 12.8 mg·min/L; P = 0.0003) (Fig. 4).

Influence of dose capping of Actinomycin D on AUC values observed in children receiving a dose of 1.5 mg/m2 (n = 32), with doses capped at 2 mg for patients with a BSA >1.3 m2 (n = 12). The box edges represent the lower (25th) and upper (75th) quartiles; the lower and upper whiskers represent the minimum and maximum AUC values observed
3.4 Actinomycin D Toxicity
Act D toxicity data were available for 146 (92 %) patients. Act D treatment was relatively well tolerated, with the most common side-effects observed being haematological toxicities. Granulocytopaenia occurred in 44 % of patients, with 35 % of patients experiencing granulocytopaenia considered Common Toxicity Criteria (CTC) grade 3 or 4. Grade 3 or 4 leucopenia and anaemia were seen in 26 and 16 % of patients respectively. Grade 3 or 4 infection (15 %), thrombocytopaenia (9 %) and fever (10 %) were also observed. Elevated alanine transaminase (ALT) and aspartate transaminase (AST) concentrations were observed in 8 and 4 % of patients respectively. However, only three cases of grade 3/4 toxicity associated with liver function were recorded. Overall, 46 patients (32 %) had no adverse events, 62 patients (43 %) had one or more grade 1/2 toxicities, and 69 patients (47 %) had one or more grade 3/4 toxicities. A summary of all toxicity data is shown in supplementary Table S3. No statistically significant associations were found between patient age, weight and BSA and grade 3 or 4 treatment-related toxicities. Analysis of Act D pharmacokinetic parameters also revealed no relationship between CL, V 1, dose or AUC and incidence of grade 3 or 4 toxicity. Any analysis of the influence of Act D pharmacokinetics on toxicity could be confounded by the co-administration of other chemotherapeutics, commonly including vincristine, ifosfamide, doxorubicin and cyclophosphamide, dependent on tumour type and stage of disease. However, no clear trends were observed for particular drug combinations in the patient population studied.
3.5 Pharmacogenetics
One hundred and forty patients provided samples for genetic analysis. Three SNPs were analyzed in ABCB1. At least one copy of the variant allele in three common exonic ABCB1 SNPs, 1236C>T, 2677G>T/A and 3435C>T were present in 44, 53 and 42 % of patients respectively. The rare ABCB1 2677A allele, resulting in a change in amino acid from serine to alanine, was present in 12 patients (allele frequency 4 %). The frequencies reported for all three SNPs were in agreement with the International HapMap Project European Caucasian population and were in Hardy–Weinberg equilibrium (supplementary Table S4).
Relationships between transporter genotype and key pharmacokinetic parameters were investigated, with valid matched pharmacokinetic and pharmacogenetic data available from 105 patients. No associations between key pharmacokinetic parameters including CL, V 1, etaCL, or etaV1 and ABCB1 genotype were observed (Fig. 5; supplementary Table S5). Equally, there was no association between genotype and treatment-related toxicity.

Effect of ABCB1 genotypes 1236C>T (a), 3435C>T (b) and 2677G>T/A (c) on Actinomycin D clearance in children with cancer. Lines represent the mean value for each genotype group
4 Discussion
Administration of Act D to paediatric patients over the last 50 years has helped to greatly improve 5 year survival rates in Wilms tumour, rhabdomyosarcoma and Ewings sarcoma, now standing at 85, 63 and 64 % respectively. Despite this, toxicity associated with Act D treatment remains a major concern and very few studies investigating the pharmacokinetics of Act D have been published.
A previous study from our group indicated a high degree of Act D pharmacokinetic variability in children with cancer, but had limited patient numbers and insufficient sampling beyond 6 h to allow characterisation of the appropriate pharmacokinetic model or robust estimation of pharmacokinetic parameters [11]. Also, pharmacogenetic studies were not appropriate in the small cohort of patients. We now report on a larger paediatric Act D pharmacokinetic patient population, designed to address the limitations of the previous study. A total of 158 patients were recruited on to this study over an eight year period, with pharmacokinetic data available from 117 patients after quality control checks. These patients represent a distinct patient population and do not include the patients studied in the previously published studies [11, 23]. A major goal of the current study was to ensure that pharmacokinetic samples were taken at both early (5–15 min) and late (20–26 h) time points. In this study, early pharmacokinetic samples at 5 min were provided by 13 (11 %) patients and at 15 min by 101 (86 %) patients. Full sample sets, containing both early and late time points were available from 82 (70 %) patients, compared to only 7 patients (23 %) in the previous study [11].
Based on pharmacokinetic samples obtained from 31 patients recruited to the original UK trial, a two-compartment model initially provided the best fit to the data, although it was clear that there were insufficient data to characterise a third disposition phase [11]. This deficiency was addressed to some extent by Mondick et al [23], who added an additional two patients with more prolonged pharmacokinetic sampling up to 48 h. However, parameter estimates from that study would be highly dependent upon those additional two patients. In a further elaboration, a putative ‘line contamination’ contribution to measured plasma concentration was modelled as a mono exponential decay over time [25]. This was introduced in an attempt to account for a poor fit of the model to the limited data set available, and at most explained an additional 19 % of variation in plasma concentrations at early time points. It was proposed that contamination could be eliminated by an appropriate catheter clearing procedure, such as that used in the current study.
Using these previously published Act D pharmacokinetic models as a reference, pharmacokinetic data from the 117 patients were analysed to determine an appropriate population pharmacokinetic model. In agreement with the previous models [11, 23, 25], these data demonstrated a very short period of rapid decline, followed by an intermediate phase and then a final slow terminal decline phase, indicative of a three compartment model. The initial pharmacokinetic model contained random effects on CL and V 1 however a much improved fit was obtained by incorporating fixed allometric scaling on all pharmacokinetic parameters (Model 1), similar to the previously published model of Mondick et al [23]. Although this model provided a reasonable fit at early and late time points, it was insufficiently flexible and tended to under predict plasma concentrations at time points between 4 and 6 h. The final model (Model 2) was obtained by incorporating additional random effects to increase the between-subject flexibility of the model, providing a significantly improved fit to data at all time points.
The substantially increased numbers of patients and pharmacokinetic samples in the current study provide more precisely estimated pharmacokinetic parameters and an increased accuracy of population estimates. For CL (13.9 L/h/70 kg) and V 1 (7.5 L/70 kg), population estimates from Model 2 were similar, but generally higher than those presented by Edwards et al [25]. While consideration of the ‘contamination factor’ by Edwards et al has been proposed as having a role to play in estimating reliable Act D pharmacokinetic parameters, by incorporating appropriate random effects and covariant parameters into the model, this factor is of secondary importance to the impact of markedly increased numbers of patients and plasma samples available for analysis in the current study.
Empirical Bayes estimates of individual pharmacokinetic parameters obtained from the current model confirms the previously reported large inter-individual variability in Act D pharmacokinetics in children with cancer. Median clearance was 5.3 L/h (range 1.5–23.5 L/h) with a coefficient of variation of 60 %. In contrast, the coefficient of variation for the random effect on CL was 28 %, demonstrating the large reduction in variability seen after adjusting for body size by allometric scaling and confirming our previous conclusion that body weight is a major determinant of Act D clearance [11].
In terms of the clinical relevance of the data obtained, analysis of Act D AUC values for all patients who received a dose of 1.5 mg/m2 showed a significantly lower mean AUC value for larger patients, where doses were capped at 2 mg, as compared to smaller children receiving a dose of 1.5 mg/m2. In essence, a dose cap of 2 mg with a 1.5 mg/m2 Act D dose means that any child with a SA >1.3 m2 will receive a lower equivalent dose. The findings from the current study bring into question the current clinical practice of dose capping at 2 mg in these larger patients. This is particularly the case when we consider that the standard US dose cap for Act D is set at a dose of 2.5 mg. Indeed if this increased dose cap had been utilised in UK patients in the current study, the AUC values in the larger patients would have been similar to those observed in the younger patients. The relationship between dose capping and reduced AUC observed in the current study would suggest that removal of the dose cap may lead to higher and potentially more beneficial plasma concentrations in older patients. However, clear correlations between Act D exposure and clinical response in children with cancer are currently lacking.
Several studies have demonstrated that treatment with Act D can cause life-threatening toxicity [5–7] and that toxicity risk can be correlated to patient age, weight and body size [8, 9, 11]. In the current study, Act D treatment was relatively well-tolerated. Haematological toxicities were by far the most common adverse effect suffered by patients, with CTC grade 1–4 haematological toxicities occurring in 44 % of the population. Increases in markers of hepatotoxicity including ALT and AST were rare, occurring in 8 and 4 % of patients respectively. Only three patients had CTC grade 3 or 4 elevated AST or ALT, therefore covariate comparisons were not possible. No significant correlations were observed between pharmacokinetic parameters and treatment related toxicities. In addition, no influence of body size, dose or pharmacokinetic parameters were found in a comparison of patients that experienced CTC grade 3 or 4 toxicity with those that experienced a lower level of toxicity or no toxicity at all.
Inter-individual variation in drug exposure between patients is a major issue, with the potential to significantly impact on cancer therapy. This may be particularly relevant for drugs used to treat tumours with better survival rates, where the majority of patients respond well, but a small percentage may experience unacceptable toxicity. Some of this variability may be accounted for by genetic variation in drug metabolising or transport proteins, as polymorphisms in these genes have the potential to alter pharmacokinetics and pharmacological phenotype. In vitro investigations and studies in knock-out mice have indicated a significant influence of ABCB1 on intracellular Act D drug concentrations and drug disposition [24], providing a sound rationale for hypothesising that SNPs in ABCB1 and other transporters could influence Act D pharmacokinetics.
The relatively large paediatric patient cohort included in the current study allowed us to investigate the potential influence of ABCB1 genotype on the pharmacokinetics of Act D. These studies were limited to the ABCB1 genotype due to the convincing supportive preclinical data indicating a role for ABCB1 in the transport of Act D [24]. The ABCB1 SNPs 1236C>T, 2677G>T/A and 3435C>T have been extensively studied with many anti-cancer agents, with the results being largely inconsistent and dependent upon drug administered and patient ethnicity. The 2677G>T/A SNP is a non-synonymous SNP, where the variant allele T results in a serine to threonine conversion or the variant allele A results in a serine to alanine conversion. Both 1236C>T and 3435C>T are synonymous SNPs, with inconsistent data concerning their effect on ABCB1 expression and on pharmacokinetics. For example lower digoxin exposure has been associated with TT genotype at position 3435 [29]. Other studies have reported better survival of glioblastoma patients with the CC genotype treated with temozolomide [30] and higher plasma concentrations of irinotecan in patients with a TT genotype at position 1236 [31]. In addition breast cancer patients who were heterozygous for each SNP had higher overall doxorubicin exposure, whilst those patients who were homozygous wild-type for all three SNPs had higher clearance in an Asian patient population [19].
In the current study, no significant association was found between ABCB1 genotype, and Act D pharmacokinetics or treatment-related toxicities. However, bearing in mind the overall patient numbers and relatively small numbers of patients in certain genotype groups (most notably the TA genotype for the 2677G>T/A polymorphism), these findings do not rule out a potential influence that might be discerned in a larger population. It is not uncommon that, despite evidence for the functional relevance of transport proteins in pre-clinical studies, both in vitro and in vivo, subsequent studies in patients fail to demonstrate any impact on pharmacokinetics. For example, the anticancer drug docetaxel has been shown in CHO cells to be a substrate for both OATP1B1 and OATP1B3 and docetaxel clearance was 18-fold lower in Oat1b2 −/− (a rodent transporter with 60 % sequence homology to human OATP1B1 and OATP1B3) mice. However, there was no effect of OAT1B1 or OAT1B3 genotype on docetaxel clearance when this was investigated in a clinical study of 141 cancer patients [32]. Our data would indicate a similar pattern in the case of Act D, with convincing data for a potential role of ABCB1 genotype obtained from pre-clinical studies, but not substantiated in a cancer patient population.
5 Conclusion
In summary we have defined for the first time the pharmacokinetics of Act D in an appropriately sized paediatric patient population, providing reliable estimates of key pharmacokinetic parameters. The current clinical practice of capping Act D doses at 2 mg in larger children in the UK is brought into question by the pharmacokinetic data obtained from this study and should be looked at in more detail. Preliminary results would also indicate that pharmacogenetic variation in ABCB1, a candidate drug transporter gene identified from in vitro and in vivo animal studies, does not significantly impact on Act D exposure in children with cancer.
References
CancerStats, Childhood Cancer—Great Britain and UK. 2010: Cancer Research UK website (http://www.cancerresearchuk.org/cancer-info/cancerstats/childhoodcancer/. Accessed 11th October, 2013).
Reich E, Franklin RM, Shatkin AJ, Tatum EL. Effect of actinomycin D on cellular nucleic acid synthesis and virus production. Science. 1961;134:556–7.
Reich E, Franklin RM, Shatkin AJ, Tatum EL. Action of actinomycin D on animal cells and viruses. Proc Natl Acad Sci USA. 1962;48:1238–45.
Goldberg IH, Rabinowitz M, Reich E. Basis of Actinomycin action II. Effect of actinomycin on the nucleotide triphosphate-inorganic pyrophosphate exchange. Proc Natl Acad Sci USA. 1962;49:226–9.
Bisogno G, de Kraker J, Weirich A, et al. Veno-occlusive disease of the liver in children treated for Wilms tumor. Med Pediatr Oncol. 1997;29:245–51.
Green DM, Finklestein JZ, Norkool P, D’Angio GJ. Severe hepatic toxicity after treatment with single-dose dactinomycin and vincristine. A report of the National Wilms’ Tumor Study. Cancer. 1988;62:270–3.
Raine J, Bowman A, Wallendszus K, Pritchard J. Hepatopathy-thrombocytopenia syndrome—a complication of dactinomycin therapy for Wilms’ tumor: a report from the United Kingdom Children’s Cancer Study Group. J Clin Oncol. 1991;9:268–73.
Arndt C, Hawkins D, Anderson JR, Brietfeld P, Womer R, Meyer W. Age is a risk factor for chemotherapy-induced hepatopathy with vincristine, dactinomycin, and cyclophosphamide. J Clin Oncol. 2004;22:1894–901.
Langholz B, Skolnik JM, Barrett JS, et al. Dactinomycin and vincristine toxicity in the treatment of childhood cancer: a retrospective study from the Children’s Oncology Group. Pediatr Blood Cancer. 2011;57:252–7.
Kanwar V, Albuquerque M, Ribeiro R, Kauffman W, Furman W. Veno-occlusive disease of liver after chemotherapy for rhabdomyosarcoma: case report with review of the literature. Med Pediatr Oncol. 1995;24:334–40.
Veal GJ, Cole M, Errington J, et al. Pharmacokinetics of dactinomycin in a pediatric patient population: a United Kingdom Children’s Cancer Study Group Study. Clin Cancer Res. 2005;11:5893–9.
Ho RH, Kim RB. Transporters and drug therapy: implications for drug disposition and disease. Clin Pharmacol Ther. 2005;78:260–77.
Melaine N, Lienard MO, Dorval I, Le Goascogne C, Lejeune H, Jegou B. Multidrug resistance genes and p-glycoprotein in the testis of the rat, mouse, Guinea pig, and human. Biol Reprod. 2002;67:1699–707.
Tatsuta T, Naito M, Oh-Hara T, Sugawara I, Tsuruo T. Functional involvement of P-glycoprotein in blood–brain barrier. J Biol Chem. 1992;267:20383–91.
Vahakangas K, Myllynen P. Drug transporters in the human blood–placental barrier. Br J Pharmacol. 2009;158:665–78.
Borst P, Elferink RO. Mammalian ABC transporters in health and disease. Annu Rev Biochem. 2002;71:537–92.
van Asperen J, van Tellingen O, Tijssen F, Schinkel AH, Beijnen JH. Increased accumulation of doxorubicin and doxorubicinol in cardiac tissue of mice lacking mdr1a P-glycoprotein. Br J Cancer. 1999;79:108–13.
Lagas JS, Fan L, Wagenaar E, et al. P-glycoprotein (P-gp/Abcb1), Abcc2, and Abcc3 determine the pharmacokinetics of etoposide. Clin Cancer Res. 2010;16:130–40.
Lal S, Wong ZW, Sandanaraj E, et al. Influence of ABCB1 and ABCG2 polymorphisms on doxorubicin disposition in Asian breast cancer patients. Cancer Sci. 2008;99:816–23.
Bray J, Sludden J, Griffin MJ, et al. Influence of pharmacogenetics on response and toxicity in breast cancer patients treated with doxorubicin and cyclophosphamide. Br J Cancer. 2010;102:1003–9.
Buda G, Ricci D, Huang CC, et al. Polymorphisms in the multiple drug resistance protein 1 and in P-glycoprotein 1 are associated with time to event outcomes in patients with advanced multiple myeloma treated with bortezomib and pegylated liposomal doxorubicin. Ann Hematol. 2010;89:1133–40.
Veal GJ, Errington J, Sludden J, et al. Determination of anti-cancer drug actinomycin D in human plasma by liquid chromatography–mass spectrometry. J Chromatogr B. 2003;795:237–43.
Mondick J, Gibiansky L, Gastonguay M, et al. Population pharmacokinetic investigation of actinomycin-D in children and young adults. J Clin Pharmacol. 2008;48:35–42.
Hill CR, Jamieson D, Thomas HD, Brown CDA, Boddy AV, Veal GJ. Characterisation of the roles of ABCB1, ABCC1, ABCC2 and ABCG2 in the transport and pharmacokinetics of actinomycin D in vitro and in vivo. Biochem Pharmacol. 2013;85:29–37.
Edwards AY, Skolnik JM, Dombrowsky E, Patel D, Barrett JS. Modeling and simulation approaches to evaluate pharmacokinetic sampling contamination from central venous catheters in pediatric pharmacokinetic studies of actinomycin-D: a report from the children’s oncology group. Cancer Chemother Pharmacol. 2012;70:83–94.
Beal S, Sheiner LB, Boeckmann A, Bauer RJ. NONMEM User’s Guides. Ellicott City: Icon Development Solutions; 1989–2009.
Lindbom L, Pihlgren P, Jonsson EN. PsN-Toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79:241–57.
Anderson BJ, Holford NH. Mechanism-based concepts of size and maturity in pharmacokinetics. Ann Rev Pharmacol Toxicol. 2008;48:303–32.
Hoffmeyer S, Burk O, von Richter O, et al. Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc Natl Acad Sci USA. 2000;97:3473–8.
Schaich M, Kestel L, Pfirrmann M, et al. A MDR1 (ABCB1) gene single nucleotide polymorphism predicts outcome of temozolomide treatment in glioblastoma patients. Ann Oncol. 2009;20:175–81.
Mathijssen RH, Marsh S, Karlsson MO, et al. Irinotecan pathway genotype analysis to predict pharmacokinetics. Clin Cancer Res. 2003;9:3246–53.
de Graan AJ, Lancaster CS, Obaidat A, et al. Influence of polymorphic OATP1B-type carriers on the disposition of docetaxel. Clin Cancer Res. 2012;18:4433–40.
Acknowledgements
This research was supported by Cancer Research UK (Grant Number C19528/A10895), the North of England Children’s Cancer Research Fund and the Experimental Cancer Medicine Centre Network. We thank the patients, research nurses and clinicians who participated in the study at the following clinical centres: Royal Victoria Infirmary, Newcastle upon Tyne; Great Ormond Street Hospital, London; Manchester Children’s Hospital; Leeds General Infirmary; Royal Liverpool Children’s Hospital; Royal Marsden Hospital, Surrey; Addenbrooke’s Hospital, Cambridge; Bristol Royal Hospital for Children; John Radcliffe Hospital, Oxford; Royal Hospital for Sick Children, Glasgow; Birmingham Children’s Hospital; Southampton General Hospital; Royal Aberdeen Children’s Hospital; Sheffield Children’s Hospital. The authors have no potential conflicts of interest to disclose.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Hill, C.R., Cole, M., Errington, J. et al. Characterisation of the Clinical Pharmacokinetics of Actinomycin D and the Influence of ABCB1 Pharmacogenetic Variation on Actinomycin D Disposition in Children with Cancer. Clin Pharmacokinet 53, 741–751 (2014). https://doi.org/10.1007/s40262-014-0153-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-014-0153-2