
Abstract
In this present study TiO2 supported metal catalysts (Pt/TiO2, Pt–Fe/TiO2) were prepared by liquid phase photo deposition method. The prepared catalysts were characterized electrochemically. X-ray Diffractometer (XRD) results showed the presence of metal particle deposition on TiO2. HR-SEM and TEM results confirmed the nano size of the metal particles and their uniform dispersion on the surface of TiO2. Electrochemical methods such as cyclic Voltammetry, linear sweep Voltammetry were used to study the stability and oxygen reduction properties of the synthesized TiO2 supported metal catalysts. The prepared catalysts were tested in Single Chamber Microbial Fuel Cell (SCMFC) with Escherichia coli of type DH5-α as bacterium. The power densities of Pt/TiO2 and Pt–Fe/TiO2 (110.5 and 136.8 mW/m2) were found to be higher compared to that of Pt/C (77 mW/m2) in MFC operation. The results visualized the better electro catalytic property for oxygen reduction in cathodic part of microbial fuel cell.
Similar content being viewed by others

Introduction
Microbial fuel cell (MFC) is a bio-electrochemical device which utilizes microorganisms and degradable organic wastes for their bioelectricity production. The working principle of MFC is similar to that of hydrogen–oxygen fuel cells [1]. In an MFC, an anaerobic anode chamber with bacterial inoculum is used in the anode to produce proton and electron from the degradation of organic wastes. The protons are transferred from the anode to the cathode through the membrane and the electrons reaches the cathode through the external circuit. The advantage of MFC technology is its simultaneous waste treatment and electricity generation. MFC technology is considered to be one of the promising alternative energy producing devices at ambient conditions [2].
A cation exchange membrane, Nafion 117® has been widely used as a polymer electrolyte membrane (PEM) in various fuel cells. However, in case of Microbial Fuel Cells (MFC), it has higher oxygen and substrate crossover from cathode to anode and vice versa respectively [3]. Extensive studies were carried out by researchers to overcome the drawbacks of Nafion and found modified polymer electrolytes such as sulfonated Poly Ether Sulfone (PES), Disulfonated poly (arylene ether sulfone) (BPSH), Sulfonated Poly Ether Ether Ketone (SPEEK) [4–7] and their composite membranes to be suitable for applications in MFC.
There are several other limiting factors that may weigh heavily against the magnitude of power yield before the MFC technology gains a significant share in the energy market. One of among the factor is sluggish oxygen reduction at cathode electrode and lower power generation as MFC is commonly governed by the cathode performance [8]. To enhance the MFC performance, high redox potential chemicals such as ferricyanide and permanganate [9, 10] were used as electron acceptors in the cathodic chamber to fasten the reduction process. A range of chemical catalysts other than Pt/C has been studied for Oxygen Reduction Reaction (ORR) in MFC, including porphyrines [11], phthalocyanines [12], MnO2 [13] and MWCNT [14]. In most MFCs, Pt/C has been used as cathode catalyst, however the carbon support increases the rate of carbon corrosion significantly and results in a decreased contact between the support and the platinum catalyst particles, which consequently becomes more mobile and forms larger catalyst particle aggregates or migrate out of the Gas Diffusion Layer (GDE) [15]. Carbon degradation can also be caused because of increased hydrophilicity of the supporting carbon, which results in deteriorated mass transport properties of the GDE [16]. In recent years, heteroatoms (Nitrogen, Boron and other similar atoms) [17–19] doped carbon materials, such as carbon blacks, graphene and carbon nanotubes, were found to exhibit excellent electro catalytic performances for ORR. Another alternate approach is using oxygen reducing bacterial organisms in cathode to replace the chemical catalyst is called bio cathodes which accepts electrons from the cathode substratum for oxygen reduction, thereby increasing the economic viability and environmentally sustainability of MFC systems. However, the comparatively lower power generation than the chemically (Pt/C) catalyzed cathode is one main downfall of bio-cathodes [20], however recent study with Ectothiorhodospiraceae bacteria showed a promising revolution towards biocathode usage in MFC [21].
Another approach is the utilization of non-precious metal catalysts. The utilization of non-precious metal alone as catalyst showed relatively less performance compared to that of Pt/C catalyst [22]. An alternate, promising approach to attain highly stable and high-efficiency ORR is the use of platinum-based alloys Pt–M (M=Co, Fe, Cu and Ni). These Pt based alloys have been demonstrated to hold higher electro catalytic activity than Pt catalyst in proton exchange membrane fuel cells [23]. The alloy catalysts have indicated the enhancement in activity as a result of the shortening of the distance to the nearest neighbor by alloying. Other properties intrinsic to Pt alloys include the exposure of a more active vicinal plane (1 0 0) on dispersed platinum particles. The exposure of the more active plane is believed to take place during the heat treatments used to induce alloy formation in the particles and explains the enhancement on the basis of the augmentation of Pt d-vacancy and the effect of higher affinity for the oxygenated species on the catalyst [24]. Pt is the best commercial catalyst we have for the ORR at present and efforts have been made to improve its activity. Alloying Pt with transition metals such as Cr, Fe, Co and Ni can improve the activity of cathodes, reduce catalyst sintering and increase the roughness of the catalyst surface and, therefore, improve performance of fuel cells with the Pt alloy cathodes [25]. Pt–Fe alloy as a cathode catalyst suggest that it contains a higher proportion of platinum active sites with a distinctly different nearest neighbor environment in relation to non-alloyed Pt sample [26, 27].
In general, non-carbon supported catalyst has been operating satisfactorily by hastening the reduction kinetics of oxygen in fuel cells for long duration which is an another method adopted to improve the cathode performance of fuel cells compared to that of commercially available carbon supported platinum (Pt/C) catalyst [28]. Using alternative materials includes semi-conductive oxides (TiOx, WOx and SnO2) or carbides [20–24, 30] in the place carbon as support are in practice. Use of this kind of semi conductive oxides/carbides promotes electro catalysis through synergistic metal-support interactions in addition to their improved corrosion resistance. However, semi-conducting oxides such as TiO2 also presents a drawback compared to carbon in that pure TiO2 is a semiconductor with a band gap of 3.2 eV for anatase and 3.0 eV for rutile [29]. Despite the lower conductivity relative to carbon, TiO2 is interesting to evaluate as catalyst support material not only due to its high stability, but also due to its better catalytic stability, activity, uniform dispersion of metal particles, metal to support interaction and favor better ORR comparable to that of carbon supported catalyst [30].
In this study, TiO2 supported Pt and Pt–Fe catalysts were prepared. The prepared Pt–Fe/TiO2 catalysts showed better and effective catalytic reaction towards ORR due to its alloy formation with Pt. The TiO2 supported catalysts have improved stability in addition to their better catalytic property compared to that of carbon supported platinum catalyst [25]. For this study Sulfonated Poly Ether Ether Ketone (SPEEK) was chosen as Polymer Electrolyte Membrane (PEM) because of its advantages over Nafion 117® and the study comprises three parts as follows, i.e., (1) the preparation of Sulfonated Poly ether ether Ketone as solid polymer electrolyte (2) Preparation of catalyst by liquid phase photo deposition method and catalyst characterization; and (3) its performance analysis in air cathode Single Chamber Microbial Fuel Cell (SCMFC).
Materials and methods
The chemicals such as PEEK (Mol. Wt. 1,00,000, Victrex), Conc. Sulfuric acid (Merck), N-Methyl Pyrollidone (NMP) (SRL), Hexachloroplatinic acid (Sigma Aldrich), TiO2 (SRL), Chloroplatinic acid (Sigma Aldrich), Ferric Chloride (Fischer Scientific, UK), Ethanol (SRL), Nutrient Media (SRL) and D (+) glucose (Sigma Aldrich) were used as such without any further purification.
Sulfonation of PEEK
PEEK polymer was sulfonated using sulfuric acid as a sulfonating agent [31]. The weighed amount of PEEK (5 g) was dissolved in concentrated sulfuric acid (90 ml) and magnetically stirred for 5 h. The reaction mixture was then poured into cold water and the sulfonated PEEK (SPEEK) was obtained in the form of white precipitate. The SPEEK was washed with deionized water for several times until the pH became neutral. The product obtained was the sulfonated form of polyether ether ketone with a sulfonation degree of 45–50 %. The SPEEK obtained from the above process was dried in a vacuum oven at 80 °C overnight. It was then dissolved in a suitable quantity of NMP (2 g in 25 ml) and cast onto a clean, dry petri dish. The membrane was obtained by evaporating the solvent in vacuum oven at 80 °C for 24 h [31]. The membrane obtained after drying was pale brown in color. It was peeled off from the petri dish and used for further analysis.
Electrocatalyst preparation
TiO2 supported metal nanocatalysts were prepared by liquid phase photo deposition method [32]. The catalysts were prepared from the aqueous solution of chloroplatinic acid with an excess of ethanol. The solution was continuously stirred and purged with nitrogen gas for 30 min to remove oxygen. The solution was irradiated with UV–Vis (365 nm) at 25 °C for 3 h using 8 numbers of 8 W Hg lamp then centrifuged and dried at 100 °C overnight. The sample was then calcinated at 450 °C for 3 h to get Pt/TiO2. The catalysts in 20 % Pt/TiO2 and Pt–Fe was taken to prepare catalyst ink for the study. Pt–Fe/TiO2 was prepared from their respective precursor in weight ratio of 1:0.5 respectively.
Characterization techniques
The prepared TiO2 supported metal nano catalysts were characterized using X-Ray Diffraction (XRD), High Resolution Scanning Electron Microscope (HR-SEM) and cyclic Voltammetry techniques.
X- ray diffraction (XRD)
The crystalline nature of the prepared catalysts was studied using XRD with a scanning rate of 2° per min. XRD measurements were performed using an X’ Pert Pro PAN analytical powder XRD instrument.
HR-SEM and TEM
A high resolution scanning electron microscopy (HR-SEM) was used to investigate the surface morphology and distribution of catalyst on the TiO2 surface. SEM micrographs were obtained with the help of Quanta 200 FEG with associated energy dispersive X-ray analyzer. The catalysts were dried and then coated with gold to achieve SEM images. Transmission Electron Microscope (TEM) images were taken using TEM Hitachi H-7500. Before analyzing the samples were dried under natural conditions.
Electrochemical measurements
The electrochemical behavior of the prepared catalyst samples was studied using Biologic VSP instrument. Glassy carbon electrode coated with catalyst was used as working electrode for each study with Saturated Calomel Electrode (SCE) and platinum as reference and counter electrodes respectively. The working electrode was prepared by taking catalyst and Vulcan XC-72 carbon and was dispersed in 1 ml of water. The suspension was ultrasonicated for 15 min. This dispersion (20 μL) was coated on glassy carbon, and 5 μL of 5 % Nafion solution (as a binder) was added to these coatings and dried. This glassy carbon was used as the working electrode. The addition of carbon was aimed at increasing the conductivity of the resultant electrode. The experiments were carried out for 200 cycles in 50 mM air-purged phosphate buffer electrolyte (pH 4.2) with a scanning rate of 50 mVs−1. LSV (Linear Sweep Voltammogram) experiments were conducted for O2 reduction reactions in the same environment with a scan rate of 10 mVs−1. All the measurements were conducted at room temperature with a oxygen saturation time of 30 min.
MFC Construction and Operation
Proton exchange membrane (SPEEK) was placed to separate the anode and cathode electrodes in SCMFC, fabricated using an acrylic cylindrical chamber of 4 cm long and 3 cm in diameter (empty bed volume of 28 ml). E coli [33–35] bacteria were used as bacterium in the anodic chamber. The anode and cathode electrodes were prepared using PTFE binder (Sigma Aldrich, India), carbon cloth (Ballard, USA) and the catalysts (Pt/C, Pt/TiO2 or Pt–Fe/TiO2) (for cathode alone) (Arora Mathey, Kolkatta) as per our previous work [36].
E coli strains (DH5-α) from our pre adapted laboratory culture collection were used and the bacterial culture was enriched by purging nitrogen gas and was kept in shaker for 48 h. The anode chamber was filled with the enriched E. coli nutrient medium and the chamber was continuously flushed with N2/CO2 (80:20) to maintain anaerobic conditions as well as the pH of the growth medium at 7. The bacterial inoculum in the anodic chamber was changed over 3–5 times (i.e., over 72–120 h) for bacterial enrichment and to allow a biofilm to form on the anode electrode surface. The chamber was refilled each time when the voltage reached a minimum value.
Polarization curve helps to understand the performance of MFC in terms of power generation. It represents the cell voltage and power density as a function of the current density. Initially, the open circuit voltage (OCV) was measured when no current was drawn. Then, the cell voltage and current were continuously monitored and recorded using a precision digital multimeter (Model 702, Metravi, India). The circuit was completed with a resistor of 1 kΩ except when different resistors (100 ohms to 1000 ohms) were used to determine the power generation as a function of load. The power density values were obtained after stable voltage was obtained. The current was calculated as
Using Ohm’s law where, I (mA) is the current, V (mV) is the voltage and R is the external resistance (Ω). Power (P) was calculated as, P = IV. The power density and current density values were calculated with respect to the area of the membrane (same for anode and cathode) used for the study which was then converted into meter square area.
Results and discussion
Properties of the membrane
The properties of SPEEK membrane were studied in detail by many researchers and the properties such as Ion exchange capacity, proton conductivity, water absorption and dissolved oxygen crossover of the membrane are listed in Table 1. The SPEEK membrane was tested in MFC and showed good results compared to that of commercial Nafion membrane [36].
XRD
X-Ray diffraction spectra of Pt/C, Pt/TiO2, Pt- Fe/TiO2 are illustrated in Fig. 1. The broad peak located at 23° 2θ accounts for carbon support for Pt/C catalyst. The spectra illustrate the presence of metals supported on TiO2. The characteristic diffraction peaks of the face centred cubic structure are detected in the platinum supported carbon catalyst samples. The diffraction peaks at 2θ values of 40, 47 and 67 are associated with the Pt (1 1 1), (2 0 0) and (2 2 0) planes, respectively and there appeared no predominant peak induced by ferric or ferric oxides in Pt–Fe/TiO2. The Pt–Fe/TiO2 presents the shift of diffraction peaks toward higher 2θ values in comparison with Pt/TiO2 catalyst, indicating the contraction of platinum lattice and formation of Pt–Fe alloy [22].

XRD patterns of a Pt/C, b Pt/TiO2 and c Pt–Fe/TiO2
Morphological studies
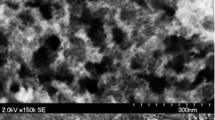
HR-SEM of the prepared catalysts is illustrated in Fig. 2. The images show the uniform dispersion of metal particles on the surface of titania without much agglomeration. The shape of the metal particle is uniform and evenly distributed on titania. Figure 2 showed the EDX data of the prepared catalyst samples. The data showed the presence of metal particles with TiO2. The SEM image reveals that the catalyst powders are made of less than 50 nm homogenous agglomerates adhered by Pt–Fe alloy particles on nano scale (Fig. 2). TEM images showed the presence of metal particles on the metal oxide support and the presence of metal particle was also observed from the Fig. 2.

HR-SEM, TEM and EDX images of a Pt/TiO2, b Pt–Fe/TiO2 catalysts
The cyclic voltammetry results of TiO2 supported catalyst were tested by three electrode method performed using Standard Calomel Electrode (SCE) as reference, Pt wire as auxiliary electrode and TiO2 catalyst coated glassy carbon electrode as working electrode using electrochemical impedance spectrometer (Biologics VSP, France). Figure 3 shows the CVs for Pt/C, Pt/TiO2 and Pt–Fe/TiO2 catalysts obtained at room temperature during continuous cycling between −0.2 and 1.2 V for a total of 200 cycles at a scan rate of 50 mV/S. The CV curves of all the prepared catalysts (Pt/C, Pt/TiO2 and Pt–Fe/TiO2) in Fig. 3 show the usual voltammogram in acid electrolytes and no additional current peak was observed, indicating that the TiO2 was electrochemically inert and functioned as a good support under operating conditions. The electro catalysts exhibited a reduction in the hydrogen under-potential adsorption/deposition area with respect to repeated potential cycling due to the dissolution of Pt and growth/re-deposition of Pt particles. The electrochemical surface areas (ECSAs) were calculated from the hydrogen desorption peaks and the same for Pt/C, Pt/TiO2 and Pt–Fe/TiO2 after the 5th cycle was found to be 87.75, 76.8 and 57.9 m2/g respectively. The ECSAs of Pt/C, Pt/TiO2 and Pt–Fe/TiO2 after 200 cycles were found to be 65.4, 69.7 and 53.1 m2/g respectively. As seen from the figure, the catalysts exhibited a decrease in ECSA with increased cycle numbers. Since, the catalysts were tested at acidic pH, the carbon support gets corroded and resulted in decreased catalyst active surface area due to catalyst particle sintering. The percentages of ECSA loss were 26, 9 and 8 % for Pt/C, Pt/TiO2 and Pt–Fe/TiO2 respectively [37]. This loss in ECSA may be due to the Ostwald ripening, Pt particle migration and sintering [38]. In contrast to carbon supported catalyst, TiO2 supported catalysts showed a comparatively smaller loss in ECSA showing higher electrochemical stability than the Pt/C.

Cyclic Voltammetry of a Pt/C, b Pt/TiO2, c Pt–Fe/TiO2 over 200 cycles
The charges in the H desorption or adsorption region are integrated to generate the number of adsorbed H atoms. Hydrogen adsorption/desorption charges on cyclic voltammograms are commonly used in the determination of Pt and Pt based nanocatalyst’s ECSAs. The charge required for either the adsorption, or desorption of a monolayer of hydrogen on a polycrystalline Pt surface is 210 μC/cm2. ECSA values can be calculated from the coulombic charge (Q H) collected in the hydrogen adsorption/desorption region from the electroactive Pt surface according to the following formula:
where, [Pt] represents the platinum loading (g cm−2) in the electrode, Q H is the charge for hydrogen desorption (mC cm−2), and 0.21 represents the charge required to oxidize a monolayer of H2 on Pt (mC cm−2).
LSV technique was used to demonstrate the oxygen reduction reactions of the catalyst [27, 39, 40]. In this study, LSV results showed the effect of electrolytes atmosphere on oxygen reduction characteristics of Pt/C, Pt/TiO2 and Pt–Fe/TiO2. Figure 4 shows the reduction peaks of the prepared catalyst in oxygen atmosphere. The catalytic activity of Pt/C, Pt/TiO2 and Pt–Fe/TiO2 was clearly observed in reduction peaks at ~0.4 V respectively. The reduction peaks of Pt/TiO2 and Pt–Fe/TiO2 were also more intense than Pt/C [39].

LSV of Pt/C, Pt/TiO2, Pt–Fe/TiO2 in O2 atmosphere
LSVs for the prepared catalysts in O2 saturated atmosphere are shown in Fig. 4. Under O2 atmosphere, TiO2 supported Pt–Fe metal alloy catalyst showed higher ORR peak current compared to Pt/TiO2, Pt/C metal catalysts, indicating that TiO2 supported Pt–Fe metal catalysts have better ORR [39].
Performance of MFC with synthesized cathode catalysts
The efficiency of the prepared cathode catalysts (Pt/TiO2 and Pt–Fe/TiO2) together with commercial Pt/C catalyst was tested in the fabricated SCMFC [41]. The performances of the SCMFCs were monitored and recorded under the same conditions. The obtained polarization curves are presented in Fig. 5. The open circuit voltages were 0.768, 0.771 and 0.778 V for the MFC using Pt/C, Pt/TiO2 and Pt–Fe/TiO2 as catalysts, respectively. The MFC using Pt–Fe/TiO2 as catalyst showed the highest voltage (0.778), higher than that of MFC using Pt/C as a catalyst (0.758 mV). To evaluate the real scale performance of the prepared catalysts, MFC with the Pt/C, Pt/TiO2 and Pt–Fe/TiO2 as the cathode catalysts was tested and the power density versus current density curves were obtained as presented in Fig. 5. The maximum power density achieved from MFCs with the Pt–Fe/TiO2 and Pt/TiO2 were 136.8 and 110.5 mW/m2, respectively, which were higher than that of MFCs with commercial Pt/C (77 mW/m2). The Pt–Fe/TiO2, Pt/TiO2 and Pt/C showed a current density of 400, 250 and 300 mA/m2 at maximum power density respectively. In this study, Pt–Fe/TiO2 showed the highest ORR activity and MFC results were consistent with the above-mentioned electrochemical properties. The better performance of TiO2 supported catalyst was due to the more corrosion resistance metal oxide support, higher electrochemical surface area of the support and the strong interaction of the support with the catalysts which were evidenced from CV and LSV results. However, in the case of iron doped Pt, the improved ORR activity associated with Pt based alloys in fuel cells are resulted due to (1) the presence of ferric composition may include the decrease of Pt–Pt bond length [42], (2) the increase of electron density in 5d orbital vacancy and the formation of stronger Pt–O2 − bonds [43], (3) the increased active site originating from the decrease of oxides on Pt surface [44].

Polarization and power density curves of SPEEK membrane with Pt/C, Pt/TiO2 and Pt–Fe/TiO2 cathode catalysts
It is noted that the anode electrode potentials were almost the same for different MFCs and not varied much with different cathode catalyst as shown in Fig. 6. While on the other hand, the cathode electrode potential varied widely and showed the same trend as the power density curves. These results suggest that the cathode catalyst material was responsible for the distinction in power production from these MFCs and the effect of the microbial populations on the anode can be ignored [45]. Variation in cathode potentials mainly resulted from the catalytic activity of the catalysts used in the cathodes towards oxygen reduction. Hence, it can be concluded that the performance of MFC can be significantly improved by using Pt–Fe/TiO2 as the most effective catalyst among the three types. This was consistent with the electrochemical results that revealed the highest catalytic activity for Pt–Fe/TiO2 among the three.

Individual Electrode potential of anode and cathode
Conclusion
A wide range of various size and shape carbon materials were used as support materials for microbial fuel cell operation. However the problem of carbon corrosion or carbon oxidation during fuel cell operation is a major drawback of carbon support. To overcome this, in this study TiO2 supported bimetallic Pt–Fe catalyst has been investigated as cathode catalyst for the oxygen electro-reduction process in single chamber microbial fuel cell and compared with a Pt/C catalyst. An improvement in the SCMFC single cell performance was observed in the presence of the Pt–Fe catalyst, due to the enhancement of the oxygen reduction kinetics. The results obtained from cyclic voltammogram experiment, LSV and MFC tests showed that the Pt–Fe/TiO2 alloy has superior catalytic activity towards oxygen reduction compared to that of Pt/C and Pt/TiO2 supported catalyst. The cyclic voltammetry experiments and ECSA calculations showed that the better stability of TiO2 supported catalysts after 200 cycles which was higher compared to that of carbon supported catalyst was due to the better interaction between metal and the support material. A maximum power density of 136.8 mW/m2 was generated from the SCMFC with Pt–Fe/TiO2 catalyst and was higher than that of Pt/C catalyst (77 mW/m2). With the capability of catalyzing ORR at low Pt utilization, the Pt–Fe alloy catalyst is expected to serve as a high-efficiency and cost-effective catalyst for sustainable recovery of electricity from organic matters with the help of MFC technology.
References
Szollosi, A., Nguyen, Q., Kovacs, A., Fogarasi, A., Kun, S., Vecseri, B.: Production of low or non-alcoholic beer in microbial fuel cell. Food Bioprod. Proc. 98, 196–200 (2016)
Yan, Z., Wang, M., Liu, J., Liu, R., Zhao, J.: Glycerol-stabilized NaBH4 reduction at room-temperature for the synthesis of a carbon-supported PtxFe alloy with superior oxygen reduction activity for a microbial fuel cell. Electrochim. Acta 141, 331–339 (2014)
Kondaveeti, S., Lee, J., Kakarla, R., Kim, H.S., Min, B.: Low-cost separators for enhanced power production and field application of microbial fuel cells (MFCs). Electrochim. Acta 132, 434–440 (2014)
Rahimnejad, M., Ghasemi, M., Najafpour, G.D., Ismail, M., Mohammad, A.W., Ghoreyshi, A.A., Hassan, S.H.: Synthesis, characterization and application studies of self-made Fe3O4/PES nanocomposite membranes in microbial fuel cell. Electrochim. Acta 85, 700–706 (2012)
Choi, T.H., Won, Y.B., Lee, J.W., Shin, D.W., Lee, Y.M., Kim, M., Park, H.B.: Electrochemical performance of microbial fuel cells based on disulfonated poly (arylene ether sulfone) membranes. J. Power Sources 220, 269–279 (2012)
Xing, P., Robertson, G.P., Guiver, M.D., Mikhailenko, S.D., Wang, K., Kaliaguine, S.: Synthesis and characterization of sulfonated poly (ether ether ketone) for proton exchange membranes. J. Membr. Sci. 229(1), 95–106 (2004)
Ayyaru, S., Dharmalingam, S.: Development of MFC using sulphonated polyether ether ketone (SPEEK) membrane for electricity generation from waste water. Bioresour. Technol. 102, 11167–11171 (2011)
Rismani-Yazdi, H., Carver, S.M., Christy, A.D., Tuovinen, O.H.: Cathodic limitations in microbial fuel cells: an overview. J. Power Sources 180(2), 683–694 (2008)
Wei, L., Han, H., Shen, J.: Effects of cathodic electron acceptors and potassium ferricyanide concentrations on the performance of microbial fuel cell. Int. J. Hydrogen Energy 37(17), 12980–12986 (2012)
You, S., Zhao, Q., Zhang, J., Jiang, J., Zhao, S.: A microbial fuel cell using permanganate as the cathodic electron acceptor. J. Power Sources 162(2), 1409–1415 (2006)
Cheng, S., Liu, H., Logan, B.E.: Power densities using different cathode catalysts (Pt and CoTMPP) and polymer binders (Nafion and PTFE) in single chamber microbial fuel cells. Environ. Sci. Technol. 40(1), 364–369 (2006)
Yuan, Y., Zhou, S., Zhuang, L.: Polypyrrole/carbon black composite as a novel oxygen reduction catalyst for microbial fuel cells. J. Power Sources 195(11), 3490–3493 (2010)
Kien, B.L., Wan, R.W.D., Mostafa, G., Kee, S., Manal, I., Swee Su, L., Jun Xing, L.: Manganese oxide/functionalised carbon nanotubes nanocomposite as catalyst for oxygen reduction reaction in microbial fuel cell. Int J Hydrogen Energy. 40, 11625–11632 (2015)
Elnaz, H., Alireza, K., Mostafa, G., Noordin, Y., Rasoul, G., Ahmad Fauzi, I.: Production of sustainable energy by carbon nanotube/platinum catalyst in microbial fuel cell. 12th Global Conference on Sustainable Manufacturing—Emerging Potentials. 26, 473–476 (2015)
Maass, S., Finsterwalder, F., Frank, G., Hartmann, R., Merten, C.: Carbon support oxidation in PEM fuel cell cathodes. J. Power Sources 176(2), 444–451 (2008)
Shao, Y., Liu, J., Wang, Y., Lin, Y.: Novel catalyst support materials for PEM fuel cells: current status and future prospects. J. Mater. Chem. 19(1), 46–59 (2009)
Kim, D.W., Li, O.L., Saito, N.: Enhancement of ORR catalytic activity by multiple heteroatom-doped carbon materials. Phys. Chem. Chem. Phys. 17(1), 407–413 (2015)
Zhi, Y., Huagui, N., Xi’an, C., Xiaohua, C., Shaoming, H.: Recent progress in doped carbon nanomaterials as effective cathode catalysts for fuel cell oxygen reduction reaction. J. Power Sources 236, 238–249 (2013)
Zhaoyan, G., Guangyuan, R., Congcong, J., Xianyong, L., Ying, Z., Lei, J., Liming, D.: High performance heteroatoms quaternary-doped carbon catalysts derived from shewanella bacteria for oxygen reduction. Sci. Rep. 5, 17064 (1–10) (2015)
Huang, L., Regan, J.M., Quan, X.: Electron transfer mechanisms, new applications, and performance of biocathode microbial fuel cells. Bioresour. Technol. 102(1), 316–323 (2011)
Quéméner, E., Rimboud, M., Bridier, A., Madigou, C., Erable, B., Bergel, A., Bouchez, T.: Biocathodes reducing oxygen at high potential select biofilms dominated by Ectothiorhodospiraceae populations harboring a specific association of genes. Bioresour. Technol. 214, 55–62 (2016)
Zhang, J.N., You, S.J., Yuan, Y.X., Zhao, Q.L., Zhang, G.D.: Efficient electrocatalysis of cathodic oxygen reduction with Pt–Fe alloy catalyst in microbial fuel cell. Electrochem. Commun. 13(9), 903–905 (2011)
Xiong, L., Kannan, A.M., Manthiram, A.: Pt–M (M¼Fe, Co, Ni and Cu) electrocatalysts synthesized by an aqueous route for proton exchange membrane fuel cells. Electrochem. Commun. 4, 898–903 (2002)
Colón-Mercado, H.R., Popov, B.N.: Stability of platinum based alloy cathode catalysts in PEM fuel cells. J. Power Sources 155(2), 253–263 (2006)
Yuan, W., Scott, K., Cheng, H.: Fabrication and evaluation of Pt–Fe alloys as methanol tolerant cathode materials for direct methanol fuel cells. J. Power Sources 163(1), 323–329 (2006)
Xu, J., Hua, K., Sun, G., Wang, C., Lv, X., Wang, Y.: Electrooxidation of methanol on carbon nanotubes supported Pt–Fe alloy electrode. Electrochem. Commun. 8(6), 982–986 (2006)
Nie, Y, Li, Land Wei, Z.: Recent advancements in Pt and Pt-free catalysts for oxygen reduction reaction. Chem. Soc. Rev. 44, 2168–2201 (2015)
Shao, Y., Yin, G., Gao, Y.: Understanding and approaches for the durability issues of Pt-based catalysts for PEM fuel cell. J. Power Sources 171(2), 558–566 (2007)
Jinfeng, Z., Peng, Z., Jianjun, L., Jiaguo, Y.: New understanding of the difference of photocatalytic activity among anatase, rutile and brookite TiO2. Phys. Chem. Chem. Phys. 16, 20382–20386 (2014)
Wang, Y.J., Wilkinson, D.P., Zhang, J.: Noncarbon support materials for polymer electrolyte membrane fuel cell electrocatalysts. Chem. Rev. 111(12), 7625–7651 (2011)
Guhan, S., Sangeetha, D.: Evaluation of sulfonated poly (ether ether ketone) silicotungstic acid composite membranes for fuel cell applications. Int. J. Polym. Mater. 58(2), 87–98 (2008)
Camacho, R., Huerta, R.G., Valenzuela, M.A., Alonso-Vante, N.: Preparation and characterization of Pt/C and Pt/TiO2 electrocatalysts by liquid phase photodeposition. Top. Catal. 54(8–9), 512–518 (2011)
Zhang, T., Cui, C., Chen, S., Ai, X., Yang, H., Shen, P., Peng, Z.: A novel mediatorless microbial fuel cell based on direct biocatalysis of Escherichia coli. Chem. Commun. 21, 2257–2259 (2006)
Zhang, T., Zeng, Y., Chen, S., Ai, X., Yang, H.: Improved performances of E. coli-catalyzed microbial fuel cells with composite graphite/PTFE anodes. Electrochem. Commun. 9, 349 (2007)
Zhang, T., Zeng, Y., Chen, S., Ai, X., Yang, H.: Improved performances of E. coli-catalyzed microbial fuel cells with composite graphite/PTFE anodes. Electrochem. Commun. 9(3), 349–353 (2007)
Prabhu, N.V., Sangeetha, D.: Characterization and performance study of sulfonated poly ether ether ketone/Fe3O4 nano composite membrane as electrolyte for microbial fuel cell. Chem. Eng. J. 243, 564–571 (2014)
Ermete, A.: Iron-containing platinum-based catalysts as cathode and anode materials for low-temperature acidic fuel cells: a review. RSC Adv. (2015). doi:10.1039/C5RA22035A
Huang, S.Y., Ganesan, P., Popov, B.N.: Titania supported platinum catalyst with high electrocatalytic activity and stability for polymer electrolyte membrane fuel cell. Appl. Catal. B 102(1), 71–77 (2011)
Shanmugam, S., Gedanken, A.: Synthesis and electrochemical oxygen reduction of platinum nanoparticles supported on mesoporous TiO2. J. Phys. Chem. C 113, 18707–18712 (2009)
Jintao, Z., Liming, D.: Heteroatom-doped graphitic carbon catalysts for efficient electrocatalysis of oxygen reduction reaction. ACS Catalysis 5(12), 7244–7253 (2015)
Liu, H., Logan, B.E.: Electricity generation using an air-cathode single chamber microbial fuel cell in the presence and absence of a proton exchange membrane. Environ. Sci. Technol. 38, 4040–4046 (2004)
Arico, A.S., Shukla, A.K., Kim, H., Park, S., Min, M., Antonucci, V.: An XPS study on oxidation states of Pt and its alloys with Co and Cr and its relevance to electroreduction of oxygen. App. Surf. Sci. 172(1), 33–40 (2001)
Mukerjee, S., Srinivasan, S., Soriaga, M.P., McBreen, J.: Effect of preparation conditions of Pt alloys on their electronic, structural, and electrocatalytic activities for oxygen reduction-XRD, XAS, and electrochemical studies. J. Phys. Chem. 99(13), 4577–4589 (1995)
Toda, T., Igarashi, H., Watanabe, M.: Enhancement of the electrocatalytic O 2 reduction on Pt–Fe alloys. J. Electroanal. Chem. 460(1), 258–262 (1999)
Zhang, J., You, S., Yuan, Y., Zhao, Q., Zhang, G.: Efficient electrocatalysis of cathodic oxygen reduction with Pt–Fe alloy catalyst in microbial fuel cell. Electrochem. Commun. 13, 903–905 (2011)
Acknowledgments
The authors thank the Department of Science and Technology (DST) India, for their financial support to carry out this work vide letter no. DST/TSG/AF/2010/09, dt. 01-10-2010.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Narayanaswamy Venkatesan, P., Dharmalingam, S. Synthesis and characterization of Pt, Pt–Fe/TiO2 cathode catalysts and its evaluation in microbial fuel cell. Mater Renew Sustain Energy 5, 11 (2016). https://doi.org/10.1007/s40243-016-0074-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40243-016-0074-0