
Abstract
Pristine multiwalled carbon nanotube supported platinum catalyst (Pt/CNT) has been developed by reduction of Pt salt on CNTs in ethylene glycol solution. The reactions were carried out under different pH conditions followed by detailed physical and electrochemical characterizations. The I–V performance of unit Polymer electrolyte membrane fuel cell shows a peak Power density of 325 mWcm−2 with catalyst prepared in alkaline medium, an increase of >250 % as compared to 87 mWcm−2 obtained, while employing catalyst prepared in acidic medium and tested under similar conditions. This is attributed not only to the uniform distribution of Pt nanoparticles with small particle size in range of 2–3 nm, but also to the metallic or zero oxidation state in which most of the Pt is present when reduction is carried out in alkaline medium.
Similar content being viewed by others
Introduction
Fuel cells are considered as the clean power delivery devices for the future that produces electricity by the electrochemical redox reactions [1–3]. It is assembled as a stack of identical repeating unit cells comprising membrane electrode assembly (MEA) in which hydrogen is oxidized at the anode and oxygen is reduced at the MEA cathode, all compressed by bi-polar plates that introduce gaseous reactants and coolants to the MEA and harvest the electric current. The cathode oxygen reduction reaction (ORR) and anode hydrogen oxidation reaction (HOR) both occur on the surfaces of Pt-based catalysts with pure water and heat as the only by-products. The catalyst performance is, therefore, very decisive to the fuel cell functioning and much effort has been guided towards its optimization [4–6]. Cost and durability are other major concerns in the commercialization of fuel cells. Platinum is not only costly, but may either be contaminated by impurities from reactant gases and/or may agglomerate at fuel cell operating temperatures. Hence, Pt on conductive supports has been proposed [7, 8]. The most commonly and commercially used support is the high surface area carbon black (Vulcan XC-72), which in principle meets the criterion of performance and cost. But in terms of durability, its performance is unsatisfactory under high temperature and humidity, low pH and strong oxidizing and reducing atmospheres [8, 9]. Its amorphous nature may cause the platinum to lose its catalytic activity due to sintering, or migration on the carbon support and subsequent dissolution into the electrolyte. A number of other catalyst supports have, therefore, been investigated [10, 11]. In the above scenario, carbon nanotubes (CNTs) prove to be a much promising alternate as catalyst support. Synthesis methods have improved over the years to produce economically viable, size-controlled CNTs in macroscopic quantities for commercial applications. CNTs would impact future science and technology since it is a highly potential material with unique structure, high crystallinity, conductivity, high accessible surface area and resistance to corrosion [12]. For prolong fuel cell operation or at high current densities, fuel starvation increases with water retention [13–17]. Teflon addition to overcome the problem decreases the conductivity. CNTs, however, have an inherent hydrophobic nature that can significantly reduce the mass transport problems.
Although research is being carried out to investigate the role of CNTs as catalyst support, there is always a scope for further improvement. Various treatments including sonochemical [18], electrochemical [19], microwave heating [20–22], and reflux heating [23] have been proposed to incorporate Pt nanoparticles onto the CNT surface. Lee et al. [24] reviewed the recent developments in the synthesis of the Pt electrocatalyst supported on CNTs for PEM fuel cell. Studies with various types of solvents [25], different reducing agents [25, 26], and varying amount of Pt [18, 26] are present in the open literature.
The present study demonstrates the development of Pt/CNT by reduction of Chloroplatinic acid on MWCNTs under different pH conditions, thus providing a detailed account of how the varying conditions of pH during the reduction process affect the properties of the synthesized nanocomposites and their corresponding behavior as a catalyst for PEM fuel cell. The effect of pH has been studied previously with high Pt loading (nearly 40 %) on CNTs. It was observed that Pt loading decreased with increasing alkalinity of the solution [23, 27]. Such high loadings are not recommended with CNTs [28, 29]. The authors have, therefore, reduced the loading to 20 % to overcome the above problem. Moreover, pristine MWCNTs have been used that not only reduces the synthesis step, but also prevents depletion of CNTs due to chemical modification. Ethylene glycol is used as a dispersing medium for CNTs which also generates reducing species in situ at high temperatures. Refluxing not only maintains the water content and pH value of the solution, but also allows uniform heating that leads to the formation of homogeneous nucleation centers [23] which act as sites for platinum deposition [30, 31]. The study is supported by exhaustive characterization. X-ray diffraction (XRD), Thermo gravimetric analysis (TGA), Transmission electron microscopy (TEM) and X-ray photoelectron spectroscopy (XPS) have been carried to determine platinum loading, structure, particle size, and oxidation states of platinum nanoparticles. Raman analysis has been carried out to evaluate the presence of defects and the interaction between Pt nanoparticles (NPs) and hexagonal carbon sheet. Electrochemical characterizations like cyclic voltammetry (CV), Linear scan voltammetry (LSV) for HOR and ORR, respectively, and unit PEM fuel cell performance have been carried out to elucidate the catalytic performance of the prepared samples.
Materials and methods
Development of Pt/CNTs
Carbon nanotube supported platinum (Pt/CNTs) samples were prepared using commercial grade Nanocyl 7000 multiwalled carbon nanotubes (MWCNTs) with diameter in the range of 20–30 nm and aspect ratio >1,000. These were uniformly dispersed in ethylene glycol (EG, 99 % pure obtained from Merck Ltd.) by ultra-sonication. Solution of 0.01 M H2PtCl6 (obtained from ACROS Organics GR grade 99 % pure) in isopropyl alcohol was added to the above solution drop by drop with constant magnetic stirring such that the ratio of Pt: CNT is 1: 4. The pH of the above solution was measured and was found to be less than 2. To prepare samples in different pH mediums 0.1 M NaOH was added to adjust the pH to 2, 7, and 11. These were further refluxed at 140 °C for 3 h in air. The process of reduction immobilizes Pt on CNTs forming Pt/CNT nanocomposites. The solution was filtered followed by washing with copious amount of de-ionized water. The filtrate was further dried to obtain powdered catalyst. The schematic of the above process is shown in Fig. 1. Dilute solution of NaBH4 was added to the residue (of the filtrate) to detect the presence of platinum. The samples prepared by refluxing the solution in acidic (pH 2), neutral (pH 7), and alkaline mediums (pH 11) have been designated as E1, E2, and E3, respectively, in the following text.

Schematic of the process of catalyst development showing a dispersion of MWCNTs in EG, b addition of Platinic acid and pH adjustment, c refluxing, d filtration and washing, and e prepared catalyst
Characterization techniques
The structural details were obtained by X-ray diffraction examination. XRD of the catalyst samples was performed on Rikagu powder X-ray diffractometer model: XRG 2KW using Cu Kα radiation. The mean crystallite size and lattice parameters were calculated from line broadening and d-spacing using Debye–Scherrer formula. The morphological studies of the prepared Pt/CNT nanocomposites were carried out with the help of TEM using Tecnai G2 F30 S-Twin instrument. For TEM analysis, samples were sonochemically dispersed in ethanol and the solution was drop casted on copper grid. Raman spectroscopy was carried out using a Renishaw InVia Reflex Micro Raman Spectrometer equipped with CCD detector at room temperature and in air. Green laser (excitation line 514 nm) was used to excite the samples. Five scans per sample were recorded wherein the samples were exposed to the laser power of 25 mW for 10 s. XPS was carried out to determine the chemical states of platinum and carbon in Pt/CNTs samples using instrument PHI Model 5000 Series. TGA was carried out on TGA/DSC 1600 by Mettler Toledo. The experiments were carried out in air from room temperature to 1,000 °C at the rate of 10 °C min−1.
Electrochemical characterization
The electrochemical measurements were performed using Biologic instrument, VSP model with EC-Lab software. Conventional three-electrode system was used for CV measurements with saturated calomel electrode (SCE), Pt and test samples as reference, counter, and working electrodes, respectively. 0.5 M HClO4 was used as electrolyte. Kinetic activity was measured ex situ by LSV in rotating disk electrode (RDE) apparatus with 5 mV/s sweep rate and 1,600 rpm speed of the rotating disk.
For fuel cell performance, Toray carbon paper samples of size 25 cm2 were teflonized and gas diffusion layer (GDL) was prepared by coating 1.5 mg/cm2 of carbon black (Vulcan XC-500) followed by sintering at 350 °C for 30 min. The prepared Pt/CNT samples (E1, E2 and E3) were coated onto the GDL layer such that the amount of Pt was maintained at 0.3 mg/cm2. Nafion Membrane-1135 (obtained from DuPont) was sandwiched in between the two electrodes (as prepared above) and hot pressed at 120–130 °C for 3 min to make the membrane electrode assembly (MEA). Fuel cell was evaluated at 60 °C and 100 % humidified conditions. Measurements of cell potential with varying current densities were conducted using electronic load procured from Bitrode Instruments, US (model: LCN4-25-24/LCN 50-24). The voltage corresponding to the applied current was measured at an interval of 30 s. Hydrogen and oxygen streams were supplied to the anode and cathode at atmospheric pressures, respectively.
Results and discussion
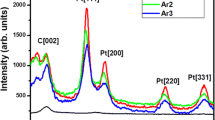
Figure 2 shows the XRD curves for pristine CNTs and Pt/CNT samples. Diffraction peak at 25.7° is attributed to graphite crystallographic planes (002) of CNTs. Pt/CNT samples show diffraction peaks at nearly 39.9°, 46.3° and 67.7°, corresponding to face-centered cubic structure of platinum. No peaks of Co or Ni are obtained which suggests that these catalyst impurities are very less. Table 1 gives a detail account of the miller indices, d-spacing and crystallite size for the given diffraction angle for samples E1, E2 and E3. The FWHM values of the samples increase with increase in pH of the synthesizing medium and correspondingly the average crystallite size decreases. The crystallite size was found to be minimum (~3 nm) for sample E3.

X-ray diffraction curves of pristine CNTs and sample E1, E2, and E3
This can be explained by the following mechanism of the reduction of Chloroplatinic acid as a function of pH. In acidic solution with low pH it is present as [PtCl6]2− and [PtCl6−x(H2O) x ](2−x)− [32].
With addition of external OH− ions in form of NaOH, there may be replacement of H2O or Cl− from the aqua species [PtCl6−x(H2O) x ](2−x) formed in step (1).
Chen et al. [33] from Extended X-ray absorption fine structure studies have confirmed that Pt–O bond length (2.03 Å) is smaller than Pt–Cl bond length (2.33 Å). Hence, due to steric contraction effect and electrostatic charge effect, the species formed in alkaline medium are smaller in size than complexes formed in acidic medium [26]. This implies that the size of platinum species deposited on the carbon support greatly depends on the pH of the environment.
The results were further confirmed by the TEM studies. Figure 3 shows the TEM images of samples E1 and E3. TEM micrographs reveal that Pt NPs are attached to the outer walls of CNTs. For sample E1, agglomeration of platinum particles is observed at some places as shown in Fig. 3a–c and the Pt particle size distribution shows maximum particles in the range of 4–5 nm (Fig. 3d). However, for sample E3, homogeneous distribution of Pt NPs with particle size in the range of 2–3 nm has been observed (Fig. 3e–h). The particle sizes determined by XRD are in close agreement with observed values from TEM. Figure 3b, d shows the presence of closed ends of CNT which further implies that during the synthesis process the CNTs remained highly intact and Pt NPs are deposited only on their surface.

TEM micrographs of sample E1 showing non-uniform distribution and agglomeration of catalyst particles (a–c) and sample E3 showing catalyst distribution (d); closed ends of CNTs after processing (e); and formed catalyst nanoparticles size of 2–3 nm (f)
A small size with a uniform distribution of the catalyst NPs is an important parameter affecting the fuel cell performance. However, the oxidation state is another important factor which decides the activity of the catalyst. It has been observed that platinum in metallic or zero oxidation state shows higher catalytic activity than its corresponding oxidized species [34]. XPS was used as a quantitative technique to measure the elemental composition and their oxidation states present in the samples so as to illustrate the chemical changes occurring due to different pH environments. The deconvoluted XPS curves (by fitting to pure Gaussian peaks) of samples E1, E2 and E3 for carbon (C1 s) and Platinum (Pt4f) are shown in Fig. 4. The peak of elemental carbon is obtained at 284.6 eV. Deconvoluting the carbon 1 s spectrum shows additional shoulder peaks at 285.8, 287.4, 289.1, and 290.7 eVs which corresponds to oxidized or functionalized carbon species.

XPS curves of catalyst samples E1, E2 and E3 for carbon 1 s (a, c, and e) and Platinum 4f peaks (b, d, and f)
Although the percentage of graphitic carbon in all samples is quite high, but there is visible contribution of other species also. The presence of sp3 hybridized carbon depicts the defects which were further confirmed from Raman analysis. As it is clear from the curves, the percentage of functionalized carbon decreases with increasing pH of the synthesizing medium. This is probably because acidic environment contributes more towards the formation of oxidized/functionalized carbon species.
XPS spectrum in Fig. 4b, d, f reveals two Pt peaks, 4f7/2 and 4f5/2 at ~70.90 and 74.14 eV, respectively, corresponding to Pt in metallic state [Pt(0)]. Deconvoluting the curves, however, presents us with shoulder peaks with a peak shift of nearly 1.4 eV (for 4f7/2) and 2.5 eV (for 4f5/2) corresponding to Pt–OH and Pt–O species, respectively. As it is clear from the curves, the percentage of Pt(0) increases and oxidized Pt decreases with increasing pH of the medium in which the catalyst is prepared. Thus, while in sample E1, 24.2 % Pt is present in oxidized state; it reduces to nearly 11.4 % in sample E3. This result further indicates that alkaline medium facilitates reduction of platinum. Thus, in all three Pt/CNT samples, the metallic state of Pt is predominant, but was found to be highest in E3, i.e., 88.6 %.
Pt/CNT-catalyzed cathode ORR may not be a complete four-electron reaction. There may be slight contribution from two-electron process also [35–37] and hydrogen peroxide formed in the process negatively affects the performance and stability of cathode and electrolyte membrane [38]. The presence of Pt–OH due to incomplete reduction may initiate the two-electron ORR process thus affecting the fuel cell performance.
The XPS data for the entire range have been given in Fig. 5 that shows the presence of C, O, Pt, Ni and Co.

The XPS data for the catalyst samples E1, E2, and E3
Figure 6 shows the Raman spectra of the different nanocomposite samples. Three bands: D (defect/disorder induced) band, G (graphite like) band, and G′ (second order harmonic to D) band have been identified. Also present is the small D + G band, which requires a defect for its activation [39]. The intensities of the bands were determined by the area under the spectral curve. The intensity of the G band (IG) has been used as a reference in determining the relative intensities of the D band (ID) and G′ band (IG′).

Raman spectra for pristine CNTs and catalyst samples E1, E2, E3 along with the calculated values of ID/IG and IG′/IG
The relative intensity of D band (ratio ID/IG) for pristine CNT sample is nearly 1.15, which indicates a large no. of defects. This is probably due to the small size of the nanotubes with closed ends (as also shown in the TEM images in Fig. 3b, e. The ends contain sp3 carbon due to pentagon–heptagon pairs and, therefore, contribute to the D band intensity. The incorporation of Pt NPs on the tubes further results in generating stresses in the hexagonal carbon layer that is reflected by the increase in the relative intensity of the D band.
The G′ band is attributed to the interlayer coupling. Its relative intensity increases very sightly in nanocomposite samples as compared to pristine MWCNTs, which indicates that the Pt nanoparticles are attached only on the outer wall of the nanotubes. This probably affects the coupling of only a few surface layers leaving the bulk unchanged.
Figure 7 shows the TGA curves of the Pt/CNT samples. TGA for pristine MWCNT without Platinum addition shows a residue of about 8.5 % which is probably due to the presence of oxides of metal (Co, Ni) used during the CNT synthesis process. For the Pt/CNT samples E1, E2, and E3, the residual content is nearly 31.9, 29.4, and 28.8 %, respectively, indicating that almost entire platinum (i.e., ~20 %) is reduced on the CNT surface. During synthesis of Pt/CNTs, it was observed that the residue left after filtration was colorless. Further addition of NaBH4 also did not lead to any change in color, thus indicating almost complete reduction of platinum on CNT surface. There is an increase in the residual content of the three samples, which should be ideally 28.5 %. This can be explained as follows. Pt will not undergo oxidation when heated in air; however, the oxidized species of Pt (as demonstrated by the XPS data) may result in oxide formation and lead to increase in the residual weight. As the oxidized Pt decreases with increasing pH of the medium in which the catalyst is prepared, similar trend is observed in the residue.

TGA and DTA curves of pristine CNTs and catalyst samples E1, E2, and E3
The DTG curves show decrease in the thermal stability of the prepared catalyst samples as compared to pristine MWCNTs. However, the decomposition temperature is much higher than the PEMFC operating temperature.
Figure 8 shows the cyclic voltammetry curves for Pt/CNTs samples prepared in different pH medium. CV measurements of Pt/CNTs samples were conducted in 0.5 M HClO4 solution over a scan range of −0.25 to 0.8 V and exhibit typical characteristic of crystalline Pt electrodes in the hydrogen and oxygen adsorption–desorption regions. The ECSA was calculated from the integrated charge associated with the hydrogen adsorption region which is based on a monolayer hydrogen adsorption on platinum and gives a measure of the HOR. ECSA for E1, E2 and E3 was found to be 102.7, 157.1 and 214.3 m2/g, respectively. The increase in the ESCA can be correlated to the decrease in the size of the deposited Pt NPs that leads to its effective utilization.

Cyclic voltammetry curves for catalyst samples E1, E2, and E3
The ORR kinetics of Pt/CNTs samples were investigated by LSV curves as shown in Fig. 9. From the LSV curves, the onset potential for the ORR was measured which gives potential at which the ORR initiates and was found to be highest for sample E3. High mid-wave potential for E3 further reflects low binding energy of the catalyst surface with the reactants which in turn implies fast catalytic activity as the catalyst surface is quickly available for reducing more oxygen.

Linear scan voltammograms for catalyst samples E1, E2, and E3
The half-cell results were further reflected in the performance of the unit PEM fuel cell. Figure 10 shows the comparative fuel cell performance with samples E1, E2, and E3 employed as catalyst. From the curves it is clear that there is a marked difference in the fuel cell performance that increases with increasing pH of the medium in which the catalyst is prepared. Maximum power density, i.e., 325 mWcm−2 has been achieved with sample E3 (employed as catalyst), much higher than that observed for catalyst samples E1 (87 mWcm−2) and E2 (201 mWcm−2). The obtained power density is much higher as compared to that of commercial catalyst as reported elsewhere [26, 40, 41]. Since the entire cell parameters are kept constant except the catalyst, the results have been explained by detailed analysis of the activation region of the polarization curves [42].

Comparative current–voltage performance of PEM fuel cell using samples E1, E2, and E3 as catalyst
The cell potential (E) versus current density (j) data for different catalyst systems can be analyzed by fitting the data to equation:
where Eeq is the equilibrium voltage and ΔVact is the activation overpotential, ΔVohmic is the over potential due to ohmic resistances in the cell; and ΔVdiff is the concentration polarization due to poor mass transport. We are mainly concerned with ΔVact due to slow electrode kinetics given by
The above Eq. (5) is linear of the form y = ax + b, where a (= RT/αnF) is the Tafel slope, j is the current density, jo is the exchange current density, n is the number of electrons transferred per mole of reactant, F is Faraday’s constant, and α the charge transfer coefficient represents the fraction of the overpotential assisting the reaction. Thus, slow reaction kinetics lead to an offset in open circuit potential E0 by an amount
Thus Eq. (4) can be written as:
The electrode kinetic parameters for the cells are summarized in Table 2, while the tafel plots are shown in Fig. 11. The value of j0.9 represents the current density at the cell potential of 0.9 V, known as kinetic current density, while α is calculated from the slope of the tafel plots. The reason for presenting j at a particular cell potential (0.9 V) instead of j0 is because even a slight variation in tafel slope can cause anomalous variation in the values of j0 [43, 44]. The values of charge transfer coefficient (α) and j0.9 increase for samples E1 to E3, whereas the tafel slopes of the polarization curves decrease indicating feasibility of the reaction. Similar values of the kinetic parameters have also been reported elsewhere [42, 45, 46]. The kinetics of the reaction can be explained by the probable mechanism. Addition of OH- ions (increasing basicity) along with EG has the tendency to catalyze the entire reduction reaction as shown in Fig. 12. This is because OH− ions has the tendency to release protons from EG (step 1).

Tafel plots of the electrochemical reactions with catalyst samples E1, E2, and E3. The tafel slops have been obtained by liner fitting

Plausible reaction mechanism when the reduction is carried out in alkaline mediums
The OH–(CH2)2–O− ion thus produced will bind on the CNT surface for stabilizing its charge. This will render the CNT surface hydrophilic. To confirm the above hypothesis 2 mg of each sample (E1, E2, and E3) was dispersed in 5 ml of water and ultrasonicated for 1 min. Figure 13 shows that sample E3 dissolves in water, while E1, and E2 do not. These additional OH− ions will also be available to reduce Pt which can bind with EG (forming square planar complexes) which is already on CNT surface (step 3). Thus, the reaction will become much more feasible as compared to when it is carried out in acidic medium (where it will be difficult to release proton). Further coordinate bonds will be stronger leading to stability of the catalyst formed.

Photograph with samples E1, E2 and E3 dispersed in water and ultrasonicated for 1 min, showing hydrophilicity of sample E3
Conclusions
Multiwalled carbon nanotube supported platinum nanoparticles prepared by reflux heating are smaller in size and well dispersed. During synthesis, high viscosity of ethylene glycol can effectively stabilize the diffusion of Pt nanoparticles. The catalytic activity of CNT-supported Pt nanoparticles greatly depends on the particle size and distribution and its oxidation state which in turn is largely affected by the pH of the synthesizing medium. The I–V performance of unit PEM fuel cell shows a peak power density of 325 mW/cm2 with catalyst prepared in alkaline medium, an increase of nearly 61 and 250 % as compared to that obtained while employing catalyst prepared in neutral and acidic mediums, respectively, and tested under similar conditions.
References
Haile, S.M.: Fuel cell materials and components. Acta. Mater. 5, 5981–6000 (2003)
Park, S., Vohs, J.M., Gorte, R.J.: Direct oxidation of hydrocarbons in a solid-oxide fuel cell. Lett. Nat. 404, 265–267 (2000)
Steele, B.C.H., Heinzel, A.: Materials for fuel-cell technologies. Nature 414, 345–352 (2001)
Hall, P.J., Bain, E.J.: Energy-storage technologies and electricity generation. Energy Policy 36, 4352–4355 (2008)
Wind, J., Späh, R., Kaiser, W., Böhm, G.: Metallic bipolar plates for PEM fuel cells. J. Power Sour. 105, 256–260 (2002)
Mathur, R.B., Maheshwari, P.H., Dhami, T.L., Tandon, R.P.: Characteristics of the carbon paper heat-treated to different temperatures and its influence on the performance of PEM fuel cell. Electrochim. Acta 52, 4809–4817 (2007)
Zhang, B., Chen, L.J., Ge, K.Y., Guo, Y.C., Peng, B.X.: Preparation of multiwall carbon nanotubes-supported high loading platinum for vehicular PEMFC application. Chin. Chem. Lett. 16, 1531–1534 (2005)
Zhang, W., Sherrell, P., Minett, A.I., Razal, J.M., Chen, J.: Carbon nanotube architectures as catalyst supports for proton exchange membrane fuel cells. Energy Environ. Sci. 3, 1286–1293 (2010)
Shao, Y., Yin, G., Gao, Y., Shi, P.: Durability study of Pt/C and Pt/CNTs catalysts under simulated PEM fuel cell conditions. J. Electrochem. Soc. 153, A1093–A1097 (2006)
Maruyama, J., Abe, I.: Enhancement effect of an adsorbed organic acid on oxygen reduction at various types of activated carbon loaded with platinum. J. Power Sour. 148, 1–8 (2005)
Shao, Y., Liu, J., Wang, Y., Lin, Y.: Novel catalyst support materials for PEM fuel cells: current status and future prospects. J. Mater. Chem. 19, 46–59 (2009)
Shao, Y., Yin, G., Zhang, J., Gao, Y.: Comparative investigation of the resistance to electrochemical oxidation of carbon black and carbon nanotubes in aqueous sulfuric acid solution. Electrochim. Acta 51, 5853–5857 (2006)
Steiner, N.Y., Mocotéguy, P.H., Candusso, D., Hissel, D.: A review on polymer electrolyte membrane fuel cell catalyst degradation and starvation issues: causes, consequences and diagnostic for mitigation. J. Power Sour. 194, 130–145 (2009)
Zhang, J., Wu, J., Zhang, H.: PEM fuel cell testing and diagnosis, pp. 301–305. Elsevier, Newnes (2013)
Chen, J., Siegel, J.B., Matsuura, T., Stefanopoulou, A.G.: Carbon corrosion in PEM fuel cell dead-ended anode operations. J. Electrochem. Soc. 158, B1164–B1174 (2011)
Dyer C.K., Moseley P.T., Ogumi Z., Rand D.A.J., Scrosati B.: Encyclopedia of Electrochemical Power Sources, pp. 852–854. Elsevier, Newnes Technology & Engineering (2013)
Jung, J., Park, B., Kim, J.: Durability test with fuel starvation using a Pt/CNF catalyst in PEMFC. Nanoscale Res. Lett. 7, 1–8 (2012)
Xing, Y.: Synthesis and electrochemical characterization of uniformly-dispersed high loading Pt Nanoparticles on sonochemically-treated carbon nanotubes. J. Phys. Chem. B 108, 19255–19259 (2004)
Guo, D.J., Li, H.L.: High dispersion and electrocatalytic properties of platinum on functional multi-walled carbon nanotubes. Electroanalysis 17, 869–872 (2005)
Zhang, W., Chen, J., Swiegers, G.F., Ma, Z.F., Wallace, G.G.: Microwave-assisted synthesis of Pt/CNT nanocomposite electrocatalysts for PEM fuel cells. Nanoscale 2, 282–286 (2010)
Chen, W.X., Lee, J.Y., Liu, Z.L.: Microwave-assisted synthesis of carbon supported Pt nanoparticles for fuel cell application. Chem. Commun. 21, 2588–2589 (2002)
Chen, W., Zhao, J., Lee, J.Y., Liu, Z.: Microwave heated polyol synthesis of carbon nanotubes supported Pt nanoparticles for methanol electro-oxidation. J. Mater. Chem. Phys. 91, 124–129 (2005)
Knupp, S.L., Li, W., Paschos, O., Murray, T.M., Snyder, J., Haldar, P.: The effect of experimental parameters on the synthesis of carbon nanotube/nanofiber supported platinum by polyol processing techniques. Carbon 46, 1276–1284 (2008)
Lee, K., Zhang, J., Wang, H., Wilkinson, D.P.: Progress in the synthesis of carbon nanotube- and nanofiber supported Pt electrocatalysts for PEM fuel cell catalysis. J. Appl. Electrochem. 36, 507–522 (2006)
Kim, P., Joo, J.B., Kim, W., Kim, J., Song, I.K., Yi, J.: NaBH4-assisted ethylene glycol reduction for preparation of carbon-supported Pt catalyst for methanol electro-oxidation. J. Power Sour. 160, 987–990 (2006)
Fang, B., Chaudhari, N.K., Kim, M.S., Kim, J.H., Yu, J.S.: Homogeneous deposition of platinum nanoparticles on carbon black for proton exchange membrane fuel cell. J. Am. Chem. Soc. 131, 15330–15338 (2009)
Stevens, D.A., Dahn, J.R.: Electrochemical characterization of the active surface in carbon-supported platinum electrocatalysts for PEM fuel cells. J. Electrochem. Soc. 150, A770–A775 (2003)
Oh, H.S., Oh, J.G., Kim, H.: Modification of polyol process for synthesis of highly platinum loaded platinum–carbon catalysts for fuel cells. J. Power Sour. 183, 600–603 (2008)
Oh, H.S., Oh, J.G., Hong, Y.G., Kim, H.: Investigation of carbon-supported Pt nanocatalyst preparation by the polyol process for fuel cell applications. Electrochem. Acta 52, 7278 (2007)
Bittencourt, C., Hecq, M., Felten, A., Pireaux, J.J., Ghijsen, J., Felicissimo, M.P., Rudolf, P., Kee, W.D., Tendeloo, G.V.: Platinum–carbon nanotube interaction. Chem. Phys. Lett. 462, 260–264 (2008)
Hull, R.V., Li, L., Xing, Y., Chusuei, C.C.: Pt nanoparticle binding on functionalized multiwalled carbon nanotubes. Chem. Mater. 18, 1780–1788 (2006)
Spieker, W.A., Liu, J., Miller, T.L., Kropf, A.J., Regalbuto, J.R.: An EXAFS study of the co-ordination chemistry of hydrogen hexachloroplatinate (IV) Speciation in aqueous solution. App. Catal. A Gen. 232, 219–235 (2002)
Chen, X., Chu, W., Wang, L., Wu, Z.: Geometry of Pt(IV) in H2PtCl6 aqueous solution: an X-ray absorption spectroscopic investigation. J. Mol. Struct. 920, 40–44 (2009)
Yoshida, H., Yazawa, Y., Hattori, T.: Effects of support and additive on oxidation state and activity of Pt catalyst in propane combustion. Catal. Today 87, 19–28 (2003)
Perez, J., Gonzalez, E.R., Ticianelli, E.A.: Oxygen electrocatalysis on thin porous coating rotating platinum electrodes. Electrochim. Acta 44, 1329–1339 (1998)
Lima, F.H.B., Ticianelli, E.A.: Oxygen electrocatalysis on ultra-thin porous coating rotating ring/disk platinum and platinum–cobalt electrodes in alkaline media. Electrochim. Acta 49, 4091–4099 (2004)
Rizo, R., Herrero, E., Feliu, J.M.: Oxygen reduction reaction on stepped platinum surfaces in alkaline media. Phys. Chem. Chem. Phys. 15, 15416–15425 (2013)
Wu, J., ZiYuan, X., Martin, J.J., Wang, H., Zhang, J., Shen, J., Wu, S., Merida, W.: A review of PEM fuel cell durability: degradation mechanisms and mitigation strategies. J. Power Sour. 184, 104–111 (2008)
Saito, R., Hofmann, M., Dresselhaus, G., Jorio, A., Dresselhaus, M.S.: Raman spectroscopy of graphene and carbon nanotubes. Adv. Phys. 60, 413–550 (2011)
Andersen, S.M., Borghei, M., Lund, P., Elina, Y.R., Pasanen, A., Kauppinen, E., Ruiz, V., Kauranen, P., Skou, E.M.: Durability of carbon nanofiber (CNF) & carbon nanotube (CNT) as catalyst support for proton exchange membrane fuel cells. Solid State Ion. 231, 94–101 (2013)
Gosh, A., Basu, S., Verma, A.: Graphene and functionalized graphene supported platinum catalyst for PEMFC. Fuel Cells 13, 355–363 (2013)
Maheshwari, P.H., Mathur, R.B.: Enhanced performance of PEM fuel cell using MWCNT reinforced carbon paper. RSC Adv. 4, 22324–22333 (2014)
Selvarani, G., Sahu, A.K., Choudhury, N.A., Sridhar, P., Pitchumani, S., Shukla, A.K.: A phenyl-sulfonic acid anchored carbon-supported platinum catalyst for polymer electrolyte fuel cell electrodes. Electrochim. Acta 52, 4871–4877 (2007)
Suárez-Alcántara, K., Rodríguez-Castellanos, A., Dante, R., Solorza-Feria, O.: Ru x Cr y Se z electrocatalyst for oxygen reduction in a polymer electrolyte membrane fuel cell. J. Power Sour. 157, 114–120 (2006)
O’Hayre, R.P.: Fuel cell fundamentals. In: Cha, S.W., Colella, W., Prinz, F.B. (eds.) Fuel cells mass transport, pp. 137–168. Wiley, New York (2006)
Barbir, F.: PEM fuel cells: theory and practice, pp. 33–72. Elsevier, Burlington (2005)
Acknowledgments
The authors are grateful to Prof. R.C. Budhani, Director, NPL, New Delhi for his support, encouragement and permission to publish the results. Thanks are also due to Dr. Vidyanand Singh and Dr. Govind for carrying out the TEM and XPS studies. One of the authors CG wants to thank CSIR, India for financial support. These studies have been carried out under the CSIR project entitled “Development of Advanced Materials for Next Generation Energy Efficient Devices (D-NEED)”.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under license to BioMed Central Ltd. Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Gupta, C., Maheshwari, P.H., Sasikala, S. et al. Processing of pristine carbon nanotube supported platinum as catalyst for PEM fuel cell. Mater Renew Sustain Energy 3, 36 (2014). https://doi.org/10.1007/s40243-014-0036-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40243-014-0036-3