
Abstract
Various extrinsic stresses, including hypoxia and oxidative stress, trigger the progression of diseases associated with inflammation and fibrosis via the pathogenic alteration of these stress signals. They include the HIF pathway, induced by hypoxia, and the Nrf2 pathway, induced by oxidative stress. Recent evidence emphasizes that the extrinsic stress-induced phenotypic changes are widely associated with endoplasmic reticulum (ER) stress and induction of the ER stress signal, unfolded protein response. This issue focuses on the recent insights demonstrating the link between molecular mechanisms leading to fibrosis and stress signals. In particular, we summarized the contribution of hypoxia, oxidative stress, and ER stress signals, all of which are common pathogenic pathways for the progression of chronic renal disease, to fibrotic changes, as well as the future aspects of therapeutic approaches targeting these stress pathways to fibrosis in various organs.
Similar content being viewed by others


Introduction
Tissue damage triggers both inflammatory and repair responses that, in the case of repeated or chronic injury, result in fibrosis, an excess deposition of extracellular matrices in an organ or tissue. Fibrosis is the common pathological feature of a variety of diseases, including tubulointerstitial fibrosis in the kidney, cirrhosis in the liver, pulmonary fibrosis in the lung, and myocardial fibrosis in the heart. The processes of fibrosis consists of injury to the tissue, recruitment of inflammatory cells, release of fibrogenic cytokines, and activation of collagen-producing cells. Here, we focus on the pathogenesis and potential therapeutic approaches against fibrosis in the kidney, which is the common final outcome of almost all progressive chronic kidney disease (CKD).
Effecter Cells in Kidney Fibrosis
Independently of the initial insult, any kind of repeated inflammation and tissue damage results in eventual kidney fibrosis. The effecter cells in fibrosis are myofibroblasts. The source of myofibroblasts has been a focus of intensive research, with candidates including tubular cells (epithelial–mesenchymal transition, EMT) and circulating cells (fibrocytes). However, accumulating evidence of fate-mapping suggests that pericytes are the main source of myofibroblasts in the kidney [1]. Molecular pathways that regulate detachment and transdifferentiation of pericytes into pathological myofibroblasts include Wingless/Int, ephrin, transforming growth factor (TGF)-β, platelet-derived growth factor (PDGF), and Hedgehog signaling pathways [1, 2]. When pericytes become myofibroblasts, they lose pericyte functions and capillaries become unstable with deleterious consequences for the kidney, including aggravation of inflammation and fibrosis. Interestingly, renal erythropoietin (EPO) producing cells, which are also localized in the interstitium, also become myofibroblasts and then lose their EPO-producing activity in the progression of fibrosis [3]. The decline of renal EPO production significantly accelerates the vicious cycle of loss of renal oxygen supply and hypoxic and oxidative stress, indicating the effects of EPO-producing cells in an increase in myofibroblasts.
The Link of Stress Signals and the Development and Progression of Fibrosis
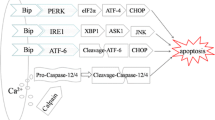
The development and progression of kidney fibrosis significantly occur when the kidney is exposed to repeated or chronic stress, not only inflammatory conditions, but also hypoxia or oxidative stress (Fig. 1). We summarize the recent reports demonstrating the link between kidney fibrosis and their stress signal pathway, as follows.

The cross talk of stress signals in the fibrotic process. It is known that fibrosis is induced by repeated or chronic inflammation. The stress signals contributing to the pathophysiology of CKD, including hypoxic, oxidative, and ER stresses, act as defensive mechanisms at an early stage of disease development. However, the recent accumulating evidence emphasizes that prolonged or overwhelming activation of these stress signals causes them to mutually affect each other, and thereby aggravate the fibrosis leading to CKD progression. Thus, HIF, Nrf2, or UPR molecules might be highlighted as novel targets for therapeutic approaches of fibrosis. CKD chronic kidney disease, ER endoplasmic reticulum, HIF hypoxia-inducible factor, Nrf2 nuclear factor-erythroid-2-related factor 2, UPR unfolded protein response
Hypoxic Stress of the Kidney
Fibrosis results in a decrease in the number of peritubular capillaries to deliver oxygen and a decrease in oxygen diffusion efficiency that is associated with the increased distance between the remaining capillaries and resident kidney cells needing oxygen, leading to hypoxia of the kidney. Hypoxia leads to aggravation of tubulointerstitial injury and fibrosis, making a vicious cycle to end-stage kidney disease (ESKD) [4, 5]. Studies utilizing blood oxygen level-dependent (BOLD)-MRI and diffusion-weighted (DW)-MRI have showed the correlation of hypoxia and fibrosis in human kidney [6].
Cells are endowed with a defensive mechanism against hypoxia, and the master regulator of adaptive responses to hypoxia is hypoxia-inducible factor (HIF). Pharmacological activation of HIF ameliorates fibrosis of the kidney in various models, including the remnant kidney and chronic progressive anti-Thy1 nephritis [7–9]. HIF activation also improves fibrosis in other organs. HIF activation promoted repair of mouse airway allograft microvasculature and attenuated fibrosis of the lung associated with chronic rejection [10]. Cardiac-specific overexpression of HIF ameliorated cardiac fibrosis associated with remodeling in diabetic mice [11].
In order to elucidate the downstream effectors of HIF in detail, we have performed studies utilizing transcriptomics, proteomics, siRNA library screening, and ChIP-seq analysis. One of the HIF target genes we identified was cytoglobin [12, 13]. Cytoglobin is a novel member of the globin superfamily and is expressed in interstitial fibroblasts of the kidney. Expression levels of cytoglobin show a compensatory increase as fibrosis of the kidney progresses. We found that cytoglobin protects the kidney against fibrosis via its anti-oxidative mechanism that is mediated by the heme domain of the molecule.
Acute kidney injury (AKI) leads to fibrosis of the kidney in the long term (for details, see “Epigenetic Changes and Fibrosis” below). Pharmacological HIF activation before AKI ameliorated fibrosis of the kidney [14]. Although fibrosis and inflammation are associated with each other in most cases, recent studies delineated a specific role of HIF in fibrosis and inflammation. Global activation of HIF suppressed inflammation and fibrogenesis in mice subjected to unilateral ureteral obstruction (UUO), whereas activation of HIF in myeloid cells suppressed inflammation of the kidney only [15]. Conversely, global deletion or myeloid-specific inactivation of HIF promoted inflammation. Studies utilizing deletion of the VEGF-A gene, a representative HIF target, in myeloid cells showed that myeloid cell-derived VEGF is essential for the prevention of fibrotic damage in the lung [16]. In contrast, studies utilizing myeloid cell-specific HIF-1 knockout mice showed that HIF activation in macrophages may promote fibrosis by regulating the production of PDGF-B in a model of liver fibrosis due to bile duct ligation [17]. Thus, a role of HIF in inflammatory cells may depend on the target organ of HIF.
Oxidative Stress of the Kidney
In CKD patients, oxidative stress is induced by multiple factors, including uremia and chronic inflammation. Priming of peripheral polymorphonuclear leukocytes is a key mediator of chronic inflammation and oxidative stress in CKD patients, occurring before the onset of renal replacement therapy and further augmented in chronic hemodialysis [18]. Intravenous administration of iron, which many CKD patients receive, also provokes the generation of bioactive iron, which enhances oxidative stress [19]. The study of 87 CKD patients with levels of plasma 8-isoprostanes (8-epiPGF2α) and serum total antioxidant status as markers of oxidative stress showed that oxidative stress increases as CKD progresses and correlates significantly with level of renal function [20]. There is growing consensus that the state of oxidative stress in CKD is correlated with the progression state of fibrosis as described below.
Nuclear factor-erythroid-2-related factor 2 (Nrf2) is the master regulator of genes that encode many antioxidant and detoxifying enzymes. Recent studies utilizing the adenine model and the remnant kidney model showed that CKD is associated with impaired Nrf2 activity, which contributes to the pathogenesis of oxidative stress, inflammation, and fibrosis [21].
To support a pathogenic role of oxidative stress in CKD, a systematic review of the Cochrane database also suggested that antioxidant therapy in pre-dialysis CKD patients may prevent progression to ESKD [22].
Oxidative stress also induces hypoxia due to a decrease in bioavailability of nitric oxide, mitochondrial uncoupling, and reduced efficiency of mitochondrial respiration [23]. Hypoxia also leads to the aggravation of oxidative stress. These two factors synergistically enhance tissue fibrosis. Indoxyl sulfate, a representative uremic toxin, induces oxidative stress and aggravates hypoxia of the kidney. Absorbents of uremic toxins ameliorate hypoxia and fibrosis of the kidney [24•]. Uremic toxins also enhance cardiac fibrosis via enhanced oxidative stress in hypertensive rats [25].
Nlandu Khodo et al. [26] demonstrated that deletion of NADPH-oxidase 4 (NOX4) is associated with increased kidney fibrosis in obstructed kidneys. This effect was associated with enhanced tubular cell apoptosis, as well as defective HIF-1 oxygen sensing and Nrf2 antioxidant pathways. Although NOX4 has been suspected to participate in kidney fibrosis, NOX4, which is expressed in tubular cells at much higher rates than podocytes or myofibroblasts, might play a different function in tubular cells and may be crucial for tubular cell survival under stressed conditions.
ER Stress at the Center of Various Stress Signals in Fibrosis
Current evidence suggests a prominent role for ER stress and activation of the UPR pathway in fibrosis. ER stress enhances fibrotic remodeling through the activation of multiple pathways, including pro-apoptotic pathways and inflammatory responses. The ER stress-inducing factors include hypoxia, oxidative stress, and inflammatory conditions [27•]. This section summarizes the impact of ER stress as well as oxidative stress in the pathophysiology of fibrosis.
The Cross talk of the Stress Signals of Hypoxia, Oxidative, and ER Stress
The ER maintains the quality of proteins by regulating the capacity for protein synthesis and folding. Under the pathogenic conditions, including oxidative stress and hypoxia, the imbalance of protein synthesis/folding capacity occurred in association with the unfolded protein accumulation in the ER, in turn leading to ER stress. The ER stress induces the stress signal, UPR. It is established that the UPR pathway links with other stress signals induced by hypoxia (HIF pathway) or oxidative stress (Nrf2 pathway), and contributes to the development/progression of various diseases, including glomerular and tubular injury leading to CKD [27•, 28].
Previous studies implicate important roles for HIF and UPR pathways in adaption to hypoxia. Interestingly, these pathways are also critical for tumor survival. HIF and eIF2α signaling of UPR contribute to the survival of hypoxic cells in vitro and in vivo [29]: eIF2α signaling induces protection against reactive oxygen species produced by hypoxia. These findings suggest that interaction between HIF and UPR under hypoxic conditions is implicated in the induction of phenotypic alteration, such as fibrosis, by hypoxia.
From the view point that Nrf2 inhibits fibrosis via suppression of both oxidative stress and inflammation, Nrf2 activation by the UPR pathway might be beneficial for the prevention of fibrosis. In particular, ER membrane-bound stress sensors, such as IRE1 and PERK, activate Nrf2. PERK phosphorylates Nrf2 and disrupts its association with Keap1, resulting in its nuclear translocation. JNK, which is activated by the IRE1 axis of UPR, also induces Nrf2–Keap1 dissociation and Nrf2 stabilization. In the nucleus, Nrf2 also upregulates genes related to the ER-associated degradation (ERAD) pathway, which eliminates the unfolded proteins from the ER and maintains ER function [30].
Evidence of the Contribution of ER Stress to Fibrosis
Due to the cross talk of stress signals described above, there is growing consensus that not only hypoxia and oxidative stress signals but also ER stress—namely, the UPR pathway—contributes to fibrosis [31, 32]. In lung fibrosis, it was demonstrated the UPR pathway depends on TGF-β induction, especially the contribution of the IRE1 or ATF6 axis on TGF-β expression [33]. Myofibroblastic differentiation of lung fibroblasts induced by TGF-β was associated with UPR activation, and a chemical chaperone, 4-phenylbutyric acid (4-PBA), suppressed TGF-β-induced UPR and profibrotic gene expression [34].
UPR activation was also observed in the tubular lesion of animal models and patients with renal fibrosis: profibrotic cytokines, TGF-β or PDGF, impaired ER function, and thereby activated the UPR pathway, i.e., activation of ER stress sensors, IRE1, ATF6, or PERK. These stress sensors aggravated ECM accumulation in the tubulointerstitium through the enhancement of the correct folding of ECM proteins by ERP57, a protein disulfide isomerase that forms a disulfide bridge for protein folding [35]. Another fibrotic phenotypic change in tubular epithelial cells, EMT, was also accelerated by UPR activation [36]. In UUO fibrosis-model rats, tubular cell apoptosis and interstitial fibrosis developed in association with proapoptotic UPR activation, and the attenuation of the overwhelming UPR activation by candesartan, which is known to modulate proapoptotic UPR, mitigated the tubulointerstitial damages [37].
Epigenetic Changes and Fibrosis
Recent epidemiological studies showed that AKI, once believed to be transient and to recover completely, often leads to development of CKD and ESKD. Mechanisms of fibrosis that develop long after an initial insult are hot topics that are intensively investigated. Renal ischemia/reperfusion of rats induced an imbalance of angiopoeitin-2/angiopoeitin-1 accompanied by the proliferation of pericytes, loss of endothelial cells and development of fibrosis 9 weeks after the initial insult [38].
An important concept for understanding the course of CKD after AKI is that the fibrotic change occurs via processes that are independent of the original insult, and it is likely that epigenetic changes of resident kidney cells induced by oxidative stress, hypoxic stress, and ER stress favor a fibrotic phenotype. These epigenetic changes include histone modifications and miRNA expressions. For example, Kato et al. [39••] reported that TGF-β, a major profibrotic molecule, increased miR-192 expression through the epigenetic modification of the miR-192 promoter region, and thereby induced renal fibrosis, such as mesangial matrix expansion in glomeruli. Overexpression of smad7 suppressed renal fibrosis via altering the renal tubular expression of not only miR-192, but also miR-21 and miR-29b in obstructive nephropathy mice [40].
We performed chromatin immunoprecipitation (ChIP) with deep sequencing (ChIP-seq) of endothelial cells exposed to hypoxia and found that HIF-1 functions as an enhancer of GLUT3 (SLC2A3) by interaction with lysine (K)-specific demethylase 3A (KDM3A). KDM3A is recruited to the GLUT3 locus in an HIF-1-dependent manner and removes a suppressive histone mark to enhance gene expression [41].
We also assessed changes in miRNA expression in the renal tubular cell line HK-2 under oxidative stress or ER stress using miRNA microarray assay. Expression of miR-205 was markedly decreased in both stress conditions, and functional analysis revealed that decreased miR-205 led to an increase in cell susceptibility to oxidative and ER stresses, and that this increase was associated with the induction of intracellular ROS and suppression of antioxidant enzymes. We found that prolyl hydroxylase 1 (PHD1), which downregulates HIF and antioxidant enzymes downstream, is a novel target of miR-205 [42].
Novel Approaches for Drug Development to Tissue Fibrosis
Therapeutic approaches that have been tried against fibrosis include pirfenidone, anti-TGF-β antibody, anti-CTGF antibody, and PAI-1 inhibitor [43, 44]. However, as described above, we have a number of potential and promising targets against fibrosis. Experimental therapies of HIF activation and ER stress-related reagents have been successful, and antioxidant treatments have been successful in humans to some extent.
Nrf2 Activators
There is growing consensus that the amelioration of pathogenic oxidative stress status by activation of the Nrf2 pathway protects the cells from fibrotic stimuli. Bardoxolone methyl, a potent inducer of Nrf2-regulated antioxidant and anti-inflammatory processes, demonstrated great promise for the therapeutic management of CKD in diabetic patients [45]. However, a larger trial in patients with CKD and type 2 diabetes was suspended in October 2012, owing to serious adverse events in the treatment arm of the investigation. Publication of more details about the adverse events is on hold because the compound received significant attention by a number of doctors and patients.
Other Nrf2 activators, which are structurally different from bardoxolone methyl, might also be promising in the prevention of not only CKD but also fibrosis-related diseases [46]. Young broccoli sprouts as a functional food contain many bioactive compounds, especially sulforaphane, which activates Nrf2; these compounds are considered as supplementary treatments in diabetes-induced fibrosis [47]. Oral sulforaphane also increased antioxidant enzymes in nasal lavage cells in humans [48]. Curcumin, a substance in turmeric, alleviates oxidative stress, inflammation, and renal fibrosis in CKD model rats by Nrf2 activation [49]. Of note, dimethyl fumarate (BG-12), another Nrf2 activator with a completely different structure from bardoxolone methyl, was shown to reduce relapse rates in patients with multiple sclerosis [50, 51].
Epigenetic Drugs
Epigenetic modification of DNA and histone play an important role in fibrosis of the lung, liver, and kidney, as described above [52–54]. Therefore, epigenetic interventions are the novel focus of intensive research, and recent studies screening for small molecules to accelerate recovery after AKI have identified the histone deacetylase (HDAC) inhibitor, methyl-4-(phenylthio)butanoate, as a promising therapeutic candidate [55]. In contrast, Sirt1, which is also an HDAC, plays a key role in the prevention of advanced glycation end product (AGE)-induced diabetic renal fibrosis. Sirt1 suppressed AGE-induced profibrotic gene expressions via activation of the Nrf2 pathway, followed by upregulation of antioxidant gene expressions in glomerular mesangial cells [56]. Of note, it was also demonstrated that advanced glycation caused alteration of Sirt1 expression [57] and epigenetic modification [58, 59], suggesting a critical role of AGE-Sirt1 interaction in diabetes-induced fibrosis via epigenetic alteration. Further investigation will open a new avenue for the development of therapeutic strategies targeting epigenetics to fibrosis.
ER Stress Modulators
Recent accumulating reports highlight the benefits of therapeutic approaches that target ER stress together with oxidative stress in kidney injury leading to fibrosis [27, 60]. Chemical chaperons, including 4-PBA and tauroursodeoxycholic acid (TUDCA), enhance protein folding capacity and restore ER function, resulting in mitigation of fibrosis in the lung and liver [34, 61, 62]. The novel effect of angiotensin II receptor blockers (ARBs; candesartan or almesartan) or anti-inflammatory drugs (mizolibine, [63]) as ER stress modulators may also be promising in prevention of fibrotic phenotypes.
ER stress is also induced by advanced glycation of protein and DNA, namely glycative stress, which is also known as the pathogenesis of glomerular sclerosis in diabetic nephropathy, and vice versa [60, 64, 65]. It establishes the vicious cycle of glycative stress and ER stress under certain pathogenic conditions, such as hyperglycemia and oxidative stress. Thus, some anti-glycation compounds can ameliorate pathogenic UPR activation, and thereby maintain cell homeostasis. The previous evidence demonstrating the renoprotective effects of ER stress modulators in glomerular and tubulointerstitial injury might support the effect of these modulators on kidney fibrosis. However, further investigations demonstrating the molecular mechanism of kidney fibrosis aggravated by ER stress are necessary for the development of novel therapeutic approaches targeting ER stress.
Conclusions
Fibrosis is a key and common phenotype for the progression of various diseases including CKD, suggesting the importance of developing more effective therapies against fibrosis to prevent disease progression. It is established that repeated or chronic inflammation triggers fibrosis. Further, hypoxic stress, oxidative stress, and ER stress also significantly trigger fibrosis by an increase in profibrotic gene expressions in various organs. The stress signal pathways, such as HIF, Nrf2, and UPR, demonstrate cross talk with each other, and some compounds used to regulate these stress pathways have already been successful in human clinical trials. HIF, Nrf2, and UPR are important targets of future therapeutic approaches against fibrotic diseases.
References
Recently published papers of particular importance have been highlighted as follows: • Of importance •• Of major importance
Ren S, Duffield JS (2013) Pericytes in kidney fibrosis. Curr Opin Nephrol Hypertens 22:471–480
Duffield JS, Lupher M, Thannickal VJ, Wynn TA (2013) Host responses in tissue repair and fibrosis. Annu Rev Pathol 8:241–276
Souma T, Yamazaki S, Moriguchi T et al (2013) Plasticity of renal erythropoietin-producing cells governs fibrosis. J Am Soc Nephrol 24:1599–1616
Nangaku M (2006) Chronic hypoxia and tubulointerstitial injury: a final common pathway to end-stage renal failure. J Am Soc Nephrol 7:17–25
Nangaku M, Inagi R, Miyata T, Fujita T (2008) Hypoxia and hypoxia-inducible factor in renal disease. Nephron Exp Nephrol 110:e1–e7
Inoue T, Kozawa E, Okada H et al (2011) Noninvasive evaluation of kidney hypoxia and fibrosis using magnetic resonance imaging. J Am Soc Nephrol 22:1429–1434
Tanaka T, Matsumoto M, Inagi R et al (2005) Induction of protective genes by cobalt ameliorates tubulointerstitial injury in the progressive Thy1 nephritis. Kidney Int 68:2714–2725
Tanaka T, Kojima I, Ohse T et al (2005) Cobalt promotes angiogenesis via hypoxia-inducible factor and protects tubulointerstitium in the remnant kidney model. Lab Invest 85:1292–1307
Song YR, You SJ, Lee YM et al (2010) Activation of hypoxia-inducible factor attenuates renal injury in rat remnant kidney. Nephrol Dial Transplant 25:77–85
Jiang X, Khan MA, Tian W et al (2011) Adenovirus-mediated HIF-1α gene transfer promotes repair of mouse airway allograft microvasculature and attenuates chronic rejection. J Clin Invest 121:2336–2349
Xue W, Cai L, Tan Y et al (2010) Cardiac-specific overexpression of HIF-1α prevents deterioration of glycolytic pathway and cardiac remodeling in streptozotocin-induced diabetic mice. Am J Pathol 177:97–105
Mimura I, Nangaku M, Nishi H et al (2010) Cytoglobin, a novel globin, plays an antifibrotic role in the kidney. Am J Physiol Renal Physiol 299:F1120–F1133
Nishi H, Inagi R, Kawada N et al (2011) Cytoglobin, a novel member of the globin family, protects kidney fibroblasts against oxidative stress under ischemic conditions. Am J Pathol 178:128–139
Kapitsinou PP, Jaffe J, Michael M et al (2012) Preischemic targeting of HIF prolyl hydroxylation inhibits fibrosis associated with acute kidney injury. Am J Physiol Renal Physiol 302:F1172–F1179
Kobayashi H, Gilbert V, Liu Q et al (2012) Myeloid cell-derived hypoxia-inducible factor attenuates inflammation in unilateral ureteral obstruction-induced kidney injury. J Immunol 188:5106–5115
Stockmann C, Kerdiles Y, Nomaksteinsky M et al (2010) Loss of myeloid cell-derived vascular endothelial growth factor accelerates fibrosis. Proc Natl Acad Sci USA 107:4329–4334
Copple BL, Kaska S, Wentling C (2012) Hypoxia-inducible factor activation in myeloid cells contributes to the development of liver fibrosis in cholestatic mice. J Pharmacol Exp Ther 341:307–316
Sela S, Shurtz-Swirski R, Cohen-Mazor M et al (2005) Primed peripheral polymorphonuclear leukocyte: a culprit underlying chronic low-grade inflammation and systemic oxidative stress in chronic kidney disease. J Am Soc Nephrol 16(8):2431–2438
Kuo KL, Hung SC, Wei YH, Tarng DC (2008) Intravenous iron exacerbates oxidative DNA damage in peripheral blood lymphocytes in chronic hemodialysis patients. J Am Soc Nephrol 19:1817–1826
Dounousi E, Papavasiliou E, Makedou A et al (2006) Oxidative stress is progressively enhanced with advancing stages of CKD. Am J Kidney Dis 48:752–760
Aminzadeh MA, Nicholas SB, Norris KC, Vaziri ND (2013) Role of impaired Nrf2 activation in the pathogenesis of oxidative stress and inflammation in chronic tubulo-interstitial nephropathy. Nephrol Dial Transplant 28(8):2038–2045
Jun M, Venkataraman V, Razavian M (2012) Antioxidants for chronic kidney disease. Cochrane Database Syst Rev 10:CD008176
Lai EY, Luo Z, Onozato ML et al (2012) Effects of the antioxidant drug tempol on renal oxygenation in mice with reduced renal mass. Am J Physiol Renal Physiol 303:F64–F74
• Palm F, Nangaku M, Fasching A (2010) Uremia induces abnormal oxygen consumption in tubules and aggravates chronic hypoxia of the kidney via oxidative stress. Am J Physiol Renal Physiol 299:F380–F386. This article highlights the impact of uremic toxins on kidney fibrosis. Indoxyl sulfate, a representative uremic toxin, promotes fibrosis via aggravation of oxidative stress and hypoxia
Yisireyili M, Shimizu H, Saito S et al (2013) Indoxyl sulfate promotes cardiac fibrosis with enhanced oxidative stress in hypertensive rats. Life Sci 92:1180–1185
Nlandu Khodo S, Dizin E, Sossauer G et al (2012) NADPH-oxidase 4 protects against kidney fibrosis during chronic renal injury. J Am Soc Nephrol 23:1967–1976
• Inagi R (2010) Endoplasmic reticulum stress as a progression factor for kidney injury. Curr Opin Pharmacol 10:156–165. This review summarizes the pathophysiological significance of cross talk of stress signaling derived from ER stress, oxidative stress, or inflammation in kidney diseases
Chiang CK, Inagi R (2010) Glomerular diseases: genetic causes and future therapeutics. Nat Rev Nephrol 6:539–554
Rouschop KM, Dubois LJ, Keulers TG et al (2013) PERK/eIF2α signaling protects therapy resistant hypoxic cells through induction of glutathione synthesis and protection against ROS. Proc Natl Acad Sci USA 110:4622–4627
Digaleh H, Kiaei M, Khodagholi F (2013) Nrf2 and Nrf1 signaling and ER stress crosstalk: implication for proteasomal degradation and autophagy. Cell Mol Life Sci. doi:10.1007/s00018-013-1409-y
Tanjore H, Lawson WE, Blackwell TS (2013) Endoplasmic reticulum stress as a pro-fibrotic stimulus. Biochim Biophys Acta 1832:940–947
Lenna S, Trojanowska M (2012) The role of endoplasmic reticulum stress and the unfolded protein response in fibrosis. Curr Opin Rheumatol 24:663–668
Maitra M, Dey M, Yuan WC et al (2013) Lung fibrosis-associated surfactant protein A1 and C variants induce latent TGF-β1 secretion in lung epithelial cells. J Biol Chem 288:27159–27171
Baek HA, Kim do S, Park HS et al (2012) Involvement of endoplasmic reticulum stress in myofibroblastic differentiation of lung fibroblasts. Am J Respir Cell Mol Biol 46:731–739
Dihazi H, Dihazi GH, Bibi A et al (2013) Secretion of ERP57 is important for extracellular matrix accumulation and progression of renal fibrosis, and is an early sign of disease onset. J Cell Sci 126:3649–3663
Dickhout JG, Carlisle RE, Austin RC (2011) Interrelationship between cardiac hypertrophy, heart failure, and chronic kidney disease: endoplasmic reticulum stress as a mediator of pathogenesis. Circ Res 108:629–642
Chiang CK, Hsu SP, Wu CT, Huang JW et al (2011) Endoplasmic reticulum stress implicated in the development of renal fibrosis. Mol Med 17:1295–1305
Khairoun M, van der Pol P, de Vries DK et al (2013) Renal ischemia-reperfusion induces a dysbalance of angiopoietins, accompanied by proliferation of pericytes and fibrosis. Am J Physiol Renal Physiol 305:F901–F910
•• Kato M, Dang V, Wang M et al (2013) TGF-β induces acetylation of chromatin and of Ets-1 to alleviate repression of miR-192 in diabetic nephropathy. Sci Signal 6:ra43. This article highlights the contribution of epigenetic alteration to kidney fibrosis in CKD with diabetes.
Chung AC, Dong Y, Yang W et al (2013) Smad7 suppresses renal fibrosis via altering expression of TGF-β/Smad3-regulated microRNAs. Mol Ther 21:388–398
Mimura I, Nangaku M, Kanki Y et al (2012) Dynamic change of chromatin conformation in response to hypoxia enhances the expression of GLUT3 (SLC2A3) by cooperative interaction of hypoxia-inducible factor 1 and KDM3A. Mol Cell Biol 32:3018–3032
Muratsu-Ikeda S, Nangaku M, Ikeda Y et al (2012) Downregulation of miR-205 modulates cell susceptibility to oxidative and endoplasmic reticulum stresses in renal tubular cells. PLoS One 7:e41462
Ha H, Oh EY, Lee HB (2009) The role of plasminogen activator inhibitor 1 in renal and cardiovascular diseases. Nat Rev Nephrol 5:203–211
Samarakoon R, Goppelt-Struebe M, Higgins PJ (2010) Linking cell structure to gene regulation: signaling events and expression controls on the model genes PAI-1 and CTGF. Cell Signal 22:1413–1419
Pergola PE, Raskin P, Toto RD et al (2011) Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med 365:327–336
Ruiz S, Pergola PE, Zager RA, Vaziri ND (2013) Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int 83:1029–1041
Bahadoran Z, Mirmiran P, Azizi F (2013) Potential efficacy of broccoli sprouts as a unique supplement for management of type 2 diabetes and its complications. J Med Food 6:375–382
Riedl MA, Saxon A, Diaz-Sanchez D (2009) Oral sulforaphane increases Phase II antioxidant enzymes in the human upper airway. Clin Immunol 130:244–2451
Soetikno V, Sari FR, Lakshmanan AP et al (2013) Curcumin alleviates oxidative stress, inflammation, and renal fibrosis in remnant kidney through the Nrf2-keap1 pathway. Mol Nutr Food Res 57:1649–1659
Gold R, Kappos L, Arnold DL et al (2012) Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med 367:1098–1107
Fox RJ, Miller DH, Phillips JT et al (2012) Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med 367:1087–1097
Bian EB, Zhao B, Huang C et al (2013) New advances of DNA methylation in liver fibrosis, with special emphasis on the cross talk between microRNAs and DNA methylation machinery. Cell Signal 25:1837–1844
Bian EB, Huang C, Wang H et al (2013) DNA methylation: new therapeutic implications for hepatic fibrosis. Cell Signal 25:355–358
Zeisberg EM, Zeisberg M (2013) The role of promoter hypermethylation in fibroblast activation and fibrogenesis. J Pathol 229:264–273
Cianciolo Cosentino C, Skrypnyk NI, Brilli LL et al (2013) Histone deacetylase inhibitor enhances recovery after AKI. J Am Soc Nephrol 24:943–953
Huang K, Huang J, Xie X et al (2013) Sirt1 resists advanced glycation end products-induced expressions of fibronectin and TGF-β1 by activating the Nrf2/ARE pathway in glomerular mesangial cells. Free Radic Biol Med 65:528–540
Cai W, Ramdas M, Zhu L et al (2012) Oral advanced glycation endproducts (AGEs) promote insulin resistance and diabetes by depleting the antioxidant defenses AGE receptor-1 and sirtuin 1. Proc Natl Acad Sci USA 109:15888–15893
Reddy MA, Natarajan R (2012) Role of epigenetic mechanisms in the vascular complications of diabetes. Subcell Biochem 61:435–454
Tonna S, El-Osta A, Cooper ME, Tikellis C (2010) Metabolic memory and diabetic nephropathy: potential role for epigenetic mechanisms. Nat Rev Nephrol 6:332–341
Inagi R (2011) Inhibitors of advanced glycation and endoplasmic reticulum stress. Methods Enzymol 491:361–380
Wang JQ, Chen X, Zhang C et al (2013) Phenylbutyric acid protects against carbon tetrachloride-induced hepatic fibrogenesis in mice. Toxicol Appl Pharmacol 266:307–316
Omura T, Asari M, Yamamoto J et al (2013) Sodium tauroursodeoxycholate prevents paraquat-induced cell death by suppressing endoplasmic reticulum stress responses in human lung epithelial A549 cells. Biochem Biophys Res Commun 432:689–694
Greenbaum LA, Benndorf R, Smoyer WE (2012) Childhood nephrotic syndrome–current and future therapies. Nat Rev Nephrol 8:445–458
Adamopoulos C, Farmaki E, Spilioti E et al (2013) Advanced glycation end-products induce endoplasmic reticulum stress in human aortic endothelial cells. Clin Chem Lab Med 2:1–10
Yamabe S, Hirose J, Uehara Y et al (2013) Intracellular accumulation of advanced glycation end products induces apoptosis via endoplasmic reticulum stress in chondrocytes. FEBS J 280:1617–1629
Compliance with Ethics Guidelines
Conflict of Interest
Reiko Inagi, Kumi Shoji, and Masaomi Nangaku declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Inagi, R., Shoji, K. & Nangaku, M. Oxidative and Endoplasmic Reticulum (ER) Stress in Tissue Fibrosis. Curr Pathobiol Rep 1, 283–289 (2013). https://doi.org/10.1007/s40139-013-0029-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40139-013-0029-4