
Abstract
Introduction
This study is part of the registrational program for intravenously administered (IV) tramadol in the USA and compared the analgesic benefit and tolerability of two doses of IV tramadol (50 mg and 25 mg) to placebo in adult patients undergoing bunionectomy, an orthopedic surgical model.
Methods
This was a phase 3, multicenter, double-blind, three-arm, randomized, placebo-controlled, multiple-dose, parallel-group trial to evaluate IV tramadol in the management of postoperative pain following bunionectomy. Eligible patients were randomized (1:1:1 ratio) to IV tramadol 50 mg, 25 mg, or placebo. Primary endpoint was summary of pain intensity differences over 48 h (SPID48). Key secondary endpoints included SPID24, total consumption of rescue analgesia, and patient global assessment of efficacy (PGA). Safety assessments included treatment emergent adverse events (TEAEs), clinical laboratory tests, vital signs, and electrocardiograms (ECGs). Assessment of the dose–response was an important objective of the study.
Results
The study established a dose response, with IV tramadol 50 mg demonstrating statistically significant benefit (p < 0.05) over placebo for primary and all key secondary efficacy endpoints, whereas tramadol 25 mg demonstrated intermediate results between the 50 mg and placebo arms. IV tramadol 50 mg was well tolerated; most common TEAEs were nausea and vomiting; and there were no meaningful differences among treatments for vital signs, ECG, and laboratory assessments. The largest proportion of patients completed tramadol 50 mg (98.6%) compared to tramadol 25 mg (91.8%) and placebo (88.2%).
Conclusion
IV tramadol 50 mg was effective and well tolerated as treatment for postoperative pain following bunionectomy surgery, while IV tramadol 25 mg, although well tolerated, was judged an ineffective dose for the treatment of pain in this setting. IV tramadol 50 mg was further developed in the registrational program for the USA.
Trial Registration
ClinicalTrials.gov identifier, NCT03290378.
Similar content being viewed by others


This was a phase 3, multicenter, double-blind, three-arm, randomized, placebo-controlled, multiple-dose, parallel-group trial. |
The study established a dose response, with IV tramadol 50 mg demonstrating statistically significant benefit (p < 0.05) over placebo for primary and all key secondary efficacy endpoints. |
IV tramadol 25 mg demonstrated numeric but not statistically significant benefit over placebo. |
IV tramadol 50 mg was well tolerated; most common were TEAEs nausea and vomiting; and there were no meaningful differences among treatments for vital signs, ECG, and laboratory assessments. |
Introduction
More than 100 million surgical procedures are performed annually in the USA, with an estimated 57.1 million performed during ambulatory surgical visits [1]. Of the 80% of patients who experience postoperative pain, fewer than half report adequate pain relief. Optimizing a patient’s pain relief in the postsurgical setting has many benefits on recovery, because poor management may contribute to medical complications as well as to the development of chronic pain [2]. Research shows that the intensity of the acute postsurgical pain correlates with the risk of developing a persistent pain state [3], suggesting that adequate postsurgical pain management is beneficial. Among patients with postsurgical pain, 1–50% develop chronic pain, depending on the type of surgery [2]. The success of inpatient and ambulatory surgery depends in part on effective postoperative pain management routines [4, 5].
Conventional opioids have been considered the mainstay of analgesia during and after surgical procedures in the USA, but questions have arisen about their use given the current opioid crisis of addiction and overdose [6]. It has become apparent that there is a need for new medicines for the management of postsurgical pain.
Intravenously administered (IV) tramadol is currently under development in the USA and may bridge the gap between non-opioid medicine and conventional opioids as an option for patients with acute pain. Tramadol is a centrally acting atypical opioid with two mechanisms of action including weak activation of the mu opioid receptor by the parent drug, more potent activation by its primary metabolite (M1), and inhibition of the reuptake of serotonin and norepinephrine. These two distinct mechanisms serve to make tramadol an effective analgesic with a good tolerability profile [7]. Tramadol is a member of the phenanthrene group of opium alkaloids, which includes morphine and codeine, and is structurally related to these opioids. Like codeine, there is a substitution of the methyl group on the phenol ring that imparts a relatively weak affinity for opioid receptors. The opioid component of tramadol comes primarily from key metabolite M1, which is a stronger mu agonist than the parent compound but has slower onset. Therefore, tramadol is generally considered a weak or atypical opioid and has been noted to have a low risk of abuse compared to conventional (i.e., Schedule II) opioids such as morphine [8, 9, 10].
Oral tramadol was approved by the US Food and Drug Administration (FDA) in 1995 and is a Schedule IV controlled substance, in contrast to many other opioids which are Schedule II or Schedule III controlled substances in the USA. The scheduling difference reflects the understanding that tramadol, as an atypical opioid with dual mechanism of action, does not carry the same abuse potential as Schedule II opioids.
It is important to note that IV tramadol is widely prescribed outside the USA (OUS) in more than 70 countries [9] but has not been approved in the USA. A novel dosing regimen for IV tramadol was recently developed for the USA, in which 50 mg is given for the first dose (T0), repeated again at 2 h and 4 h, and then once every 4 h thereafter. A phase 1 study compared this regimen to tramadol 100 mg administered orally once every 6 h (this is the highest approved oral dosage in the USA) [11]. Compared to orally administered tramadol, IV tramadol reached initial peak serum concentration (Cmax) more rapidly, while resulting in similar overall steady-state Cmax and area under the plasma concentration–time curve (AUC). It is important to note that IV tramadol as compared to orally administered tramadol results in less metabolic conversion and a slower onset of exposure to M1, the more potent mu opioid agonist than the parent compound. This is due to the avoidance of first-pass metabolism when tramadol is administered by the intravenous route.
This study was conducted following the phase 1 pharmacokinetic study and utilized the novel dosing regimen mentioned above. It was part of the registrational program for IV tramadol in the USA. The study compared the analgesic benefit and tolerability of two doses of IV tramadol (50 mg and 25 mg) to placebo in adult patients undergoing bunionectomy, an orthopedic surgical model. To our knowledge, this was the first registrational quality study that compared IV tramadol to placebo (a gold standard for US FDA approval for pain medicine) and definitively demonstrated a dose–response for IV tramadol.
Methods
Study Design and Ethics
The primary objective of this phase 3, multicenter, double-blind, three-arm, randomized, placebo-controlled, multiple-dose, parallel-group trial was to evaluate the safety, tolerability, and efficacy of two different doses of IV tramadol versus placebo in the management of postoperative pain over 48 h in patients undergoing a unilateral primary first metatarsal bunionectomy surgery. Secondary efficacy objectives included assessment of analgesic efficacy over 24 h, rescue medicine (ibuprofen 400 mg) use, and patient global assessment (PGA). An important aim of the study was dose finding, and thus two doses of tramadol were studied: 50 mg and 25 mg (each given with the same dosing intervals). Safety assessments included treatment emergent adverse events (TEAEs), clinical laboratory tests, vital signs, and electrocardiograms (ECGs).
The study was performed in accordance with ethical principles that have their origin in the Declaration of Helsinki and are consistent with International Conference on Harmonization (ICH)/Good Clinical Practice (GCP), applicable regulatory requirements, and the sponsor or its delegate’s policy on bioethics. Aspire Institutional Review Board (https://aspire-irb.com/, Santee, CA), the central IRB that approved the study for Avenue Therapeutics and the study sites, reviewed study documents according to current US regulations. The patient’s signed and dated informed consent form (ICF) was obtained before initiation of any study procedures. The study was registered at clinicaltrials.gov (NCT03290378).
Study Treatment and Eligibility
The study was conducted at five sites in the USA from August 30, 2017 to April 23, 2018. Eligible patients were male or female, age 18–75 years of age, undergoing unilateral first metatarsal bunionectomy surgery, and who met the American Society of Anesthesiologists (ASA) physical class 1 or 2, and were willing and able to understand the study procedures and use of pain scales. Important exclusion criterial included other concomitant opioids during the treatment within 30 days prior to surgery, other prohibited medications (analgesics, medications that lower seizure threshold), allergy or physical dependence on opioids, and not reaching a minimal postsurgical pain level of moderate to severe on a categorical pain scale and a numeric pain rating scale (NPRS) rating of at least 5 on a scale of 0–10.
Patients underwent the screening visit, preoperative assessment (within 24 h prior to surgery start time), the surgical/treatment visit (including the surgery), the primary treatment period of double-blind study medication for 48 h, and a follow-up visit (day 14). The treatment groups were:
-
IV tramadol 50 mg, given at hours 0, 2, 4, 8, 12, 16, 20, 24, 28, 32, 36, 40, and 44
-
IV tramadol 25 mg, given at hours 0, 2, 4, 8, 12, 16, 20, 24, 28, 32, 36, 40, and 44
Patients were confined at the healthcare facility during study drug administration and were discharged only if clinically stable. All patients in the study followed an anesthesia protocol and were not randomized into the trial if they deviated from the protocol as listed below.
Pre- and intraoperative anesthesia per study protocol included IV midazolam, a popliteal sciatic nerve block (0.5% ropivacaine; loading volume at the discretion of the investigator), a local Mayo block using short-acting 2% lidocaine without epinephrine, propofol induction followed by a continuous infusion for intraoperative sedation, intraoperative IV fentanyl, and no nausea prophylaxis is allowed. Postoperative anesthesia included regional anesthesia via a popliteal sciatic nerve block. After the completion of surgery, a continuous infusion of ropivacaine, mepivicaine, or bupivacaine was provided with additional boluses of local anesthetic at the discretion of the investigator. Additional pain medicine allowed were ketorolac 30 mg IV q6h until 6 h before removal of the popliteal block and 2–4 mg IV morphine sulfate every 2 h as needed until 1.5 h before removal of the popliteal block. Popliteal block was permanently discontinued between approximately 4:00 a.m. and 5:00 a.m. on the day after surgery.
Following removal of the popliteal block, patients were assessed for the dosing eligibility criteria; patients must have been awake and alert and had a pain intensity of 5 or greater on the 0 (no pain) to 10 (worst pain possible) point numerical pain rating scale (NPRS) and reported a score of moderate or severe on a four-point categorical rating scale (with categories of none, mild, moderate, or severe) just before the first dose of study drug. Patients who did not report pain at these levels within 8 h of the removal of the block were discontinued from the study (and thus were not treated with double-blind study medication). Bunionectomy, a common procedure used in clinical trials studying pain management, is a very painful orthopedic surgical model. The surgical anesthetic administration was the same for all patients and thus any effects on initial NPRS scores would be the same across all treatment groups. Further, postsurgical dosing criteria were included in the protocol to ensure that patients had sufficient pain levels when entering the study and therefore sufficient time for the ropivacaine or bupivacaine to wear off. The 8-h window for eligibility after removal of the block was sufficient as most patients met the criteria in that timeframe. Blinded study drug infusions were given over approximately 15 ± 2 min via an infusion pump.
We note that this study was designed using one of several well-established acute pain surgical models [12, 13]. The bunionectomy surgical pain model was not intended to “mimic” real-life treatment but rather to eliminate as many potential biases and confounding factors (e.g., from intrinsic and extrinsic factors) and to allow for the same procedures to be performed in a blinded setting, thus ensuring rigorous and reliable conclusions regarding the treatment benefit of each dose level of IV tramadol.
Endpoints
Pain intensity assessments were recorded immediately prior to the first dose (baseline, T0) and at 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 12, 14, 16, 18, 20, 22, 24, 26, 28, 30, 32, 34, 36, 38, 40, 42, 44, 46, and 48 h after first treatment (i.e., post T0). The sum of pain intensity differences (SPID) through 48 h post first dose (SPID48) measured at rest was used as the primary measure of efficacy.
Secondary efficacy endpoints include SPID24, total consumption (mg) of rescue (supplemental) analgesia (this is the total amount of rescue analgesia given to the patient after first dose of study medication through 48 h post first dose), and PGA of efficacy at 24 and 48 h post first dose using a five-point scale [the question posed was “How would you rate the study medication in terms of its effectiveness in controlling your pain?” (0 = poor; 1 = fair; 2 = good; 3 = very good; 4 = excellent)]. Additional analgesia endpoints included the time-specific pain intensity profile over time, time (in minutes) to first rescue analgesia from the time of first dose of study medication, and number (percentage) of patients who required no rescue analgesia from T0 to T48.
TEAEs, classified by the Medical Dictionary for Regulatory Activities (MedDRA) system organ class (SOC) and preferred term, including assessment of AEs related to substance abuse (based on the FDA’s guidance “Assessment of Abuse Potential of Drugs, Guidance for Industry, January 2017”). Clinical laboratory (including full chemistry, hematology, and urinalysis panels), vital sign, physical examination, and ECG changes were assessed. Clinically meaningful changes in these safety outcomes were reported as TEAEs.
Statistical Methods
The sample size was based on an assumed effect size between tramadol and placebo of at least 40%. A sample size of 135 patients per arm (405 patients in total) provided over 90% power to detect a SPID48 effect size of 40% between each pairwise tramadol and placebo comparison, assuming an alpha of 0.05 and using a two-sided test of mean differences.
The full analysis set (FAS) population was defined as all randomized patients who received at least one dose of study medication. Patients were analyzed according to the treatment group they were randomized to. The safety population was defined as all patients who received at least one dose of study medication. Patients were analyzed according to the actual treatment they received. There was one patient randomized to placebo who received a single dose of IV tramadol 50 mg, and thus was included in the tramadol 50 mg arm for purposes of safety assessment.
All SPID calculations were performed using the standard trapezoidal rule:
where PIDi is the PID at time i, and (Ti + 1 – Ti) is the time difference in hours between time i and time i + 1.
Multiple imputation provides a useful strategy for analyzing data sets with missing values. Instead of filling in a single value for each missing value, Rubin’s [14] multiple imputation strategy replaces each missing value with a set of plausible values that represent the uncertainty about the correct value to impute. This methodology was used to address missing data. Specifically, for the primary endpoint (SPID48), 100 imputed data sets were created, with data imputation for missing values due to missingness at random as well as due to discontinuation due to AE (missing, not at random) and to account for use of rescue medication (the last NPRS prior to the use of any rescue medication was used to impute subsequent NPRS scores for the subsequent protocol-specified time points for measurement of pain intensity through 4 h after the time of the dosing of the rescue medication).
The 100 imputed data sets were analyzed using an analysis of covariance (ANCOVA) model with contrasts to test the primary efficacy endpoint. The model used treatment as the main effect and investigational center and baseline pain intensity (NPRS scores of 0–10) as covariates. Data from all three treatment groups were included in the same ANCOVA model for purposes of the testing procedures. The 100 resulting treatment effect parameters and standard errors (SEs) from these ANCOVA were presented individually and then combined to provide a distribution of parameters (and SEs) upon which the primary hypothesis of treatment effect was determined.
The results of the analyses were presented in summary tables with standard summary statistics as well as least squares (LS) means, active versus placebo LS mean differences (treated group – placebo), SEs, confidence intervals, and paired-comparison p values. The mean SPID values and standard errors of the mean (SEM) for each treatment group were also presented graphically. This analysis was referred to as the multiple imputation analysis.
Average patient global assessment at scheduled time points was assessed for treatment group differences using an ANCOVA with pooled study center and the baseline pain score as covariates. Total consumption of rescue medication was calculated as the total amount of rescue analgesia (mg) captured in the rescue medications electronic case report form and recorded as given to the patient between the first dose of study medication through 48 h post first dose (4 h after the start of the last dose of study medication). The total consumption of rescue analgesia was analyzed using the nonparametric Wilcoxon rank sum test to test each active versus placebo comparison separately. The incidence of TEAEs was summarized for each treatment group by SOC and preferred term sorted in descending frequency by SOC, and then by preferred term within SOC.
There were two pairwise comparisons of interest:
-
High-dose tramadol (50 mg) versus placebo
-
Low-dose tramadol (25 mg) versus placebo
All inferential assessments were two-sided tests performed at the 0.05 alpha level. A hierarchical alpha testing strategy was utilized to control for the overall experiment-wise alpha. As there were multiple tests being performed (the single primary efficacy variable pairwise test and the key secondary efficacy tests), the following strategy was applied.
The high-dose tramadol arm versus placebo comparison was assessed for the primary endpoint, summary of pain intensity differences through 48 h (SPID48) at the 0.05 alpha level. If and only if the p value is 0.05 or less for this pairwise comparison was the lower dose tramadol arm versus placebo comparison to be assessed. If the p value for the high-dose arm was not significant, all testing was to cease, and it was to be concluded that neither tramadol treatment group provide better pain relief than placebo.
If the primary endpoint was significant for the tramadol versus placebo comparison (in favor of the tramadol arm) for the high dose comparison, then statistical testing was to proceed to the high-dose tramadol key secondary endpoints, to be tested in the following order:
-
SPID24
-
Total consumption of rescue analgesia
-
Patient global assessment of efficacy at 24 and 48 h (the two time points were to be tested simultaneously)
If a statistical test within each pairwise comparison was significant for the high dose comparison at the nominal 0.05 level, two-sided (in favor of the tramadol arm) for the first endpoint, then testing was to proceed to the next endpoint in the list, and so on. Once a non-significant test occurred, endpoints lower in the list were to be considered not statistically significant. Testing for the low-dose tramadol vs placebo comparison proceeded in a similar fashion, but only if the original high-dose tramadol vs placebo SPID48 primary comparison was statistically significant and in favor of tramadol. Dose–response was assessed via testing for a linear trend across the doses (where placebo was set to zero, and the tramadol 25 mg and 50 mg groups were analyzed accordingly).
The time-specific pain intensity profile over time was assessed using a mixed model repeated measures (MMRM) with pain intensity difference as the dependent variable and fixed effects for treatment, time, the treatment by time interaction, and random patient effect, and pooled study center and baseline pain score as covariates. Pairwise comparisons at each time point were determined from the MMRM, and model-based least square means (LS means) and standard errors (SEs) were plotted for visual comparison of the treatment group outcomes. Only scheduled visits were included in the MMRM after appropriate imputation for rescue. All analyses were performed using the SAS System® version 9.4.
A computer-generated randomization scheme was prepared prior to the study start (Axiom Real Time Metrics, Toronto, Ontario). Patients who met the enrollment criteria were randomly allocated to one of three treatment arms (tramadol 50 mg, tramadol 25 mg, or placebo) in a 1:1:1 ratio. Upon randomization, a unique four-digit study-specific randomization number was assigned. Patients, study staff, the sponsor, and contract research organizations involved with the study were blinded to treatment assignments, and the study remained blinded until all patients completed all study procedures and the clinical database had been locked.
Results
Patient Disposition
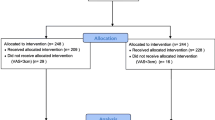
A total of 818 patients were screened, 434 were randomized, and 409 met the postoperative eligibility criteria and received treatment. Study completion rates were very high, with the largest proportion of patients completing the tramadol 50 mg group (98.6%) as compared to the tramadol 25 mg group (91.8%) and placebo group (88.2%) (Fig. 1). Discontinuation due to lack of efficacy was highest for the placebo arm (11 patients), the IV tramadol 25 mg arm (7 patients) as compared to the IV tramadol 50 mg arm (1 patient). Patients were enrolled from five investigational study centers, with each center contributing at least 12% of total patients in the study. No center contributed more than 35% of patients, and thus the influence (weight) of each study center on the outcomes was generally similar.

Study consort diagram of patient disposition from screening to study completion
Demographics
The majority of patients were female (85.3%), White (68.5%), and non-Hispanic/non-Latino (63.6%). The median age was 46.0 years and median BMI was 28 kg/m2. The treatment groups were similar with respect to the demographic and baseline characteristics (Table 1).
Primary and Secondary Efficacy
There was very little missing data for the primary efficacy endpoint: overall, 89.7% of patients had complete (i.e., no missing) NPRS assessments through hour 48. The IV tramadol 50 mg group had the highest proportion of patients with complete NPRS data (97.8%), followed by the IV tramadol 25 mg group (88.1%) and placebo group (83.1%).
The study outcomes demonstrated a clear dose response over the dose range tested (Table 2). IV tramadol 50 mg was effective in the management of postoperative pain over the full 48-h treatment period as well as over the first 24 h of treatment, whereas IV tramadol 25 mg, while having some activity, was suboptimal with respect to clinical benefit. For each efficacy parameter, the IV tramadol 50 mg group was found to be statistically significantly and clinically meaningfully better than placebo, with all p values less than 0.05 (in accordance with the predefined hierarchical testing strategy) for the primary efficacy and all key secondary efficacy endpoints. The tramadol 25 mg group was not found to be statistically significantly different from placebo for the primary analysis (SPID48), and thus testing of the key secondary findings for that dose is not presented in the table, in accordance with the predefined hierarchical testing strategy.
Pain scores were significantly lower (Fig. 2) and patient satisfaction with pain control was significantly higher with IV tramadol 50 mg as compared to placebo at both 24 and 48 h (Fig. 3), demonstrating the clinical benefit of tramadol over placebo. Patients randomized to the IV tramadol 50 mg dose used less rescue medication. The highest rate of consumption was observed in the placebo group (32.71 mg/h), followed by the tramadol 25 mg group (31.22 mg/h), and tramadol 50 mg group (21.75 mg/h). The difference in consumption between the tramadol 50 mg group and placebo group was statistically significant (p < 0.001). Patients in the IV tramadol 50 mg also experienced faster perceptible and meaningful pain relief than patients treated with placebo, and (as demonstrated via a 98.7% study completion rate) were able to tolerate the study medication well.

Least squares means (± standard errors of the means) of pain intensity differences over the 48-h treatment period (FAS population)

Patient global assessment of treatment at hour 24 and hour 48: tramadol 50 mg vs placebo
Statistically significant differences between IV tramadol 50 mg and placebo, favoring greater pain relief in the IV tramadol 50 mg group, were observed as early at the first measured time point (hour 0.5, p = 0.020) (Fig. 4) and continued to be significantly different through most of the 48-h treatment period. IV tramadol 25 mg also produced greater changes from baseline than placebo at most time points; however, statistically significant differences between IV tramadol 25 mg and placebo were only observed at two time points (hours 6 and 10). A statistically significant linear trend in key efficacy outcomes was observed across the studied dose range, i.e., placebo (0 mg), IV tramadol 25 mg, and IV tramadol 50 mg. IV tramadol 50 mg resulted in statistically significantly shorter median time to first perceptible pain relief and time to first meaningful pain relief than placebo, whereas IV tramadol 25 mg was not found to be statistically significantly different from placebo.

Least squares means (± standard errors of the means) of pain intensity differences over the first 4 h of treatment: tramadol 50 mg vs placebo (FAS population)
Safety and Tolerability
Approximately half (51.1%) of all patients experienced at least one TEAE during the study. The incidence of patients with at least one TEAE was greatest in the IV tramadol 50 mg group (66.4%), followed by the placebo group (44.1%) and the IV tramadol 25 mg group (42.1%).
The incidence of patients with at least one TEAE considered to be at least possibly related to study drug was greatest in the IV tramadol 50 mg group (58.6%), followed by the IV tramadol 25 mg group (35.3%) and placebo group (33.8%). The incidence of patients experiencing at least one TEAE of grade 3 or higher was 3.6% in the IV tramadol 50 mg group, 0.8% in the tramadol 25 mg group, and 0.7% in the placebo group. One patient who was in the IV tramadol 25 mg group experienced an SAE. Three patients (including the patient who had an SAE) had TEAEs leading to study discontinuation: two in the tramadol 25 mg group and one in the tramadol 50 mg group. No TEAEs leading to death were reported (Table 3).
Overall the types of TEAEs were similar across treatment groups, including the placebo group. The incidence of patients reporting nausea, vomiting, dizziness, and somnolence was greater for the IV tramadol 50 mg group (32.1%, 20.0%, 15.0%, and 11.4%, respectively) compared to the placebo group (8.1%, 3.7%, 2.9%, and 2.2%, respectively) and IV tramadol 25 mg group (9.0%, 3.0%, 5.3%, and 4.5%, respectively) (Table 4). The rate of patients experiencing the first onset of a TEAE in the 0–4-h time period after initiating treatment was 14.7%, 15.8%, and 28.6% in the placebo, tramadol 25 mg, and tramadol 50 mg groups, respectively. The rate of patients experiencing the first onset of a TEAE in the 4–8-h time period was 5.9%, 1.5%, and 7.1% in the placebo, tramadol 25 mg, and tramadol 50 mg groups, respectively.
While there were some patients with shifts in laboratory parameters from baseline to end of study, these shifts were not judged to be related to study treatment, and there were no trends towards a higher incidence of shifts with increasing tramadol dosage. Vital signs demonstrated a slight reduction in heart rate across the dose range, but no apparent trend in oxygen saturation (Fig. 5). Clinically meaningful changes in these safety outcomes were reported as TEAEs, with very few events reported in any treatment group.

Mean heart rate (beats per minute) and oxygen saturation (%)—change from baseline
Table 5 presents the incidence of new-onset potentially clinically significant findings that occurred at any time point during the study. The incidence of these findings was generally similar between treatment groups. Potentially clinically significant changes in QT or QTcF were only observed in the placebo group.
Discussion
This study was a phase 3, multicenter, double-blind, three-arm, randomized, placebo-controlled, multiple-dose, parallel-group trial to evaluate the safety, tolerability, and efficacy of IV tramadol 25 and 50 mg versus placebo in the management of postoperative pain in consenting patients undergoing unilateral primary first metatarsal (bunionectomy) surgery. The primary objective of this study was to evaluate the analgesic efficacy of IV tramadol compared to placebo in the management of postoperative pain. SPID48 was used as the primary measure of efficacy. Secondary efficacy endpoints included SPID24, total consumption of rescue (supplemental) analgesia, and the patient global assessment of efficacy at 24 and 48 h post first dose. Bunionectomy is a proven, reliable, and useful orthopedic model for assessing the efficacy of acute pain drugs [13].
The study outcomes demonstrate that the tramadol 50 mg dose was effective in the management of postoperative pain over both the full 48-h treatment period as well as over the first 24 h of treatment. For each efficacy parameter, the tramadol 50 mg dose was found to be statistically significantly better than placebo, with all p values less than 0.05 (in accordance with the predefined hierarchical testing strategy). Pain scores were significantly lower and patient satisfaction with pain control was significantly higher with tramadol 50 mg as compared to placebo at both 24 and 48 h. Patients randomized to the tramadol 50 mg dose used less rescue medication and experienced faster perceptible and meaningful pain relief than patients treated with placebo, and (as demonstrated via a 98.7% study completion rate) were able to tolerate the study medication well. The tramadol 25 mg was found to not be statistically significantly different from placebo in the primary analysis (SPID48), and thus formal testing of the key secondary findings was not performed for tramadol 25 mg, in accordance with the predefined hierarchical testing strategy.
Other key findings included a fast onset of action, as demonstrated by statistically significant differences between IV tramadol 50 mg and placebo as early at the first measured time point (hour 0.5, p = 0.020) and sustained through most of the 48-h treatment period (Fig. 3).
The safety assessments indicate that treatment with IV tramadol 25 mg and 50 mg was well tolerated in patients (tolerability was measured by how many patients had to discontinue treatment) with no clinically relevant changes in vital signs, laboratory tests, or ECGs. TEAEs were generally dose related, with the 25 mg group close to placebo in terms of most AEs. The incidence of at least possibly related TEAEs was highest in the tramadol 50 mg group for nausea, vomiting, dizziness, somnolence, infusion site pain, constipation, pruritus, and hypoxia. These AEs were anticipated on the basis of the pharmacology of tramadol and its primary metabolite.
This study was part of a broad 1000-patient drug development program, in which four clinical studies were performed, initiated with a phase 1 pharmacokinetics study [11] which demonstrated that the IV tramadol 50 mg dose (given with the dosing regimen as used in this bunionectomy study) provided comparable Cmax and Cmin (trough) concentrations as the oral formulation of tramadol at steady state. A phase 3 study [15] provided proof of efficacy of IV tramadol 50 mg vs both placebo and an active comparator, IV morphine, in a double-blind abdominoplasty surgical model, and importantly, demonstrated that the 50 mg dose of IV tramadol was comparable to IV morphine for both the primary and all key secondary efficacy pain parameters.
Further, a phase 3 open-label study [16] of IV tramadol 50 mg performed in various types of surgeries (e.g., knee replacement, hip replacement, soft tissue surgeries) confirmed that IV tramadol used in a multimodal setting (in which a combination of both IV tramadol and other non-opioid medications such as ibuprofen were used to treat postsurgical pain) was both effective and well tolerated in these acute pain settings.
This study, as well as the other phase 3 double-blind study (abdominoplasty surgical model), was well controlled in an established acute pain surgical model [17]. It is acknowledged that the eligibility and study procedures in such surgical model studies are not necessarily reflective of real-world practice. Procedures in these types of rigorous clinical trials must be standardized for all treatment arms and allow for detection of true treatment benefit without confounding from other intrinsic or extrinsic factors. The intent of these studies is to test primary hypotheses comparing the test therapy to a control arm without bias or confounding, and thus not necessarily to mimic real-life treatment. Rather, real-life studies (e.g., as reported by Minkowitz 2020) which are not hypothesis-testing are specifically designed to mimic a real-life setting.
In summary, this study demonstrated that IV tramadol 50 mg dosing regimen had a significant analgesic effect in patients with moderate to moderately severe pain following bunionectomy surgery. The IV tramadol 50 mg was also well tolerated in this study. Importantly, the study established a clear dose response between the 50 mg and the 25 mg dosing regimens for efficacy as well as adverse effects. The IV tramadol 50 mg dosing regimen was chosen to be taken into future clinical studies.
This study was part of an overall drug development program designed to bring IV tramadol to the USA as a treatment option. This current study was designed with a specific purpose, i.e., to identify (from one of two dosing regimens) the one with the preferred benefit–risk profile, and to establish the magnitude of any dose–response profile that may exist across the dose range studied. Thus, this study was limited in that it was not designed to answer each and every question that might relate to the benefits and risks of IV tramadol as a treatment for postsurgical pain. The broad development program (performed in over 1000 subjects in total) included phase 1 studies to ascertain pharmacokinetic concentrations from various dosing regimens and comparison to the current oral formulation of tramadol [11], assessment of the effects of tramadol on QTcF (not published), and phase 3 double-blind comparison to an active comparator [15], another phase 3 open-label “real-world” study in various types of surgery and combinations of pain relief modalities [16], i.e., multimodal use of IV tramadol as part of physician-managed pain relief across multiple study centers and types of procedures. Those studies address many of the limitations of this current study. This current study provides key information regarding dose response of IV tramadol.
Conclusion
IV tramadol 50 mg was effective and well tolerated as treatment for postoperative pain following bunionectomy surgery, while IV tramadol 25 mg, although well tolerated, was judged an ineffective dose for the treatment of pain in this setting. IV tramadol 50 mg has been further studied in a large registrational program for the USA.
References
Cullen KA, Hall MJ, Golosinskiy A. Ambulatory surgery in the United States, 2006. Natl Health Stat Report. 2009;11:1–25.
Apfelbaum JL, Chen C, Mehta SS, Gan TJ. Postoperative pain experience: results from a national survey suggest postoperative pain continues to be undermanaged. Anesth Analg. 2003;97:534–40.
Kehlet H, Jensen TS, Woolf CJ. Persistent postsurgical pain: risk factors and prevention. Lancet. 2006;367(9522):1618–25.
George NE, Gurk-Turner C, Etcheson JI, et al. The addition of diclofenac to a multimodal pain control regimen decreases postoperative pain and opioid consumption. Surg Technol Int. 2017;22:31.
Cheung CW, Wong SSC, Qiu Q, et al. Oral oxycodone for acute postoperative pain: a review of clinical trials. Pain Physician. 2017;20:SE33–52.
Brummett CM, Waljee JF, Goesling J, et al. New persistent opioid use after minor and major surgical procedures in US adults. JAMA Surg. 2017;152(6):e170504.
Grond S, Sablotzki A. Clinical pharmacology of tramadol. Clin Pharmacokinet. 2004;43:879–923.
World Health Organization (WHO). Expert committee on drug dependence. Tramadol update review report. 2014. https://www.who.int/medicines/areas/quality_safety/6_1_Update.pdf. Accessed 13 Apr 2019
Grünenthal GmBH. Application for inclusion of tramadol into the WHO Model List of Essential Medicines (EML) 2017. World Health Organization. 2017. https://www.who.int/selection_medicines/committees/expert/21/applications/Grunethal_tramadol.pdf. Accessed 13 Apr 2019.
Schneider MF, Bailey JE, Cicero TJ, et al. Integrating nine prescription opioid analgesics and/or four signal detection systems to summarize statewide prescription drug abuse in the United States in 2007. Pharmacoepidemiol Drug Saf. 2009;18(9):778–90.
Lu L, Ryan M, Harnett M, Atiee GJ, Reines SA. Comparing the pharmacokinetics of 2 novel intravenous tramadol dosing regimens to oral tramadol: a randomized 3-arm crossover study. Clin Pharmacol Drug Dev. 2020;9(4):537–46.
Singla N, Bindewald M, Singla S, et al. Efficacy and safety of intravenous meloxicam in subjects with moderate-to-severe pain following abdominoplasty. Plast Reconstr Surg Glob Open. 2018;6(6):e1846. https://doi.org/10.1097/GOX.0000000000001846.
Riff D, Duckor S, Gottlieb I, et al. Diclofenac potassium liquid-filled soft gelatin capsules in the management of patients with postbunionectomy pain: a phase III, multicenter, randomized, double-blind, placebo-controlled study conducted over 5 days. Clin Ther. 2009;31:10.
Rubin DB. Inference and missing data. Biometrika. 1976;63:581–92.
Minkowitz H, Salazar H, Leiman D, et al. Intravenous tramadol is effective in the management of postoperative pain following abdominoplasty: a three-arm randomized placebo and active-controlled trial. Drugs R&D. 2020. https://doi.org/10.1007/s40268-020-00309-0.
Minkowitz H, Leiman D, Lu L, et al. IV tramadol – a new treatment option for management of post-operative pain in the US: an open-label, single-arm, safety trial including various types of surgery. J Pain Res. 2020;13:1155–62. https://doi.org/10.2147/JPR.S251175.
Singla NK, Desjardins PJ, Chang PD. A comparison of the clinical and experimental characteristics of four acute surgical pain models: dental extraction, bunionectomy, joint replacement, and soft tissue surgery. Pain. 2014;155:441–56.
Acknowledgments
Special thanks to Robert Criscola and Amy Landry Wheeler for support and during the performance of this clinical study.
Funding
This study and the journal’s Rapid Service Fee was funded by Avenue Therapeutics.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authorship Contributions
Neil K. Singla was an investigator on the study and helped design the study. Richard Pollak, Ira Gottlieb, David Leiman, Harold Minkowitz, and John Zimmerman were investigators on the study. Mark Harnett helped design the study, performed statistical analysis and interpretation, and was responsible for drafting the manuscript. Michael Ryan was the clinical project lead on the study and interpreted the outcomes. Lucy Lu helped design the study and interpreted the outcomes. Scott Reines helped design the study and interpreted the outcomes.
Disclosures
Lucy Lu, Scott Reines, and Mike Ryan are employees of the study sponsor, Avenue Therapeutics. Neil K. Singla, Richard Pollak, Ira Gottlieb, David Leiman, Harold Minkowitz, John Zimmerman, and Mark Harnett were compensated, either directly as employees of contract research groups who were contracted for support in the performance of this study or directly as consultants to Avenue Therapeutics.
Compliance with Ethics Guidelines
The study was performed in accordance with ethical principles that have their origin in the Declaration of Helsinki and are consistent with International Conference on Harmonization (ICH)/Good Clinical Practice (GCP), applicable regulatory requirements, and the sponsor or its delegate’s policy on bioethics. Aspire Institutional Review Board (https://aspire-irb.com/, Santee, CA), the central IRB that approved the study for Avenue Therapeutics and the study sites, reviewed study documents according to current US regulations. The patient’s signed and dated informed consent form (ICF) was obtained before initiation of any study procedures. The study was registered at clinicaltrials.gov (NCT03290378).
Data Availability
The data sets generated during and/or analyzed during the current study are part of the New Drug Application under review at the US FDA and is not publicly available.
Author information
Authors and Affiliations
Corresponding author
Additional information
Digital Features
To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12588047.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Singla, N.K., Pollak, R., Gottlieb, I. et al. Efficacy and Safety of Intravenously Administered Tramadol in Patients with Moderate to Severe Pain Following Bunionectomy: A Randomized, Double-Blind, Placebo-Controlled, Dose-Finding Study. Pain Ther 9, 545–562 (2020). https://doi.org/10.1007/s40122-020-00184-2
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40122-020-00184-2