
Abstract
Historically, vitamin K antagonists have been the only class of oral anticoagulants available. Despite our experience with warfarin over the past 60 years, its use is associated with several pharmacokinetic and clinical disadvantages including unpredictable dosing, frequent monitoring, and delayed onset and offset. Edoxaban, an oral direct Xa inhibitor, may provide clinicians with an additional option in patients requiring chronic anticoagulation. This review examines the pharmacology and clinical data of edoxaban as a therapeutic alternative.
Similar content being viewed by others


Introduction
Anticoagulation is a critical component in the management of venous thromboembolism (VTE) and atrial fibrillation. Venous thromboembolism, including deep vein thrombosis (DVT) and pulmonary embolism (PE), has an annual incidence rate of approximately 1 per 1000 people in the general population. Furthermore, VTE is the third most common acute cardiovascular disorder after myocardial infarction and stroke, affecting approximately 900,000 persons in the United States annually [1, 2]. Atrial fibrillation is the most prevalent cardiac arrhythmia, affecting over 2.2 million people in the United States and 4.5 million people in the European Union [3].
Vitamin K antagonists (VKAs) have been the standard of care for the prevention and treatment of VTE, and stroke prevention in atrial fibrillation patients. Prior to 2009, the only oral anticoagulant available in the United States was warfarin. Three months of warfarin for idiopathic VTE resulted in a 95% reduction in recurrent thromboembolism [4]. Warfarin has been found to reduce the risk of stroke by 64% compared to placebo in patients with atrial fibrillation [5]. The rate of major bleeding with warfarin in the setting of venous thromboembolism has been estimated to be approximately 6.5% per year, with a range of 3% in low-risk patients to 30% in high-risk patients [6]. In the setting of atrial fibrillation, the rate of major bleeding has been estimated to be approximately 2.3% per year [7]. The limitations of VKAs are well documented, including a narrow therapeutic window that requires monitoring, variable therapeutic response, delayed onset and offset of action, dose adjustments, food and drug interactions, and risk for bleeding.
In 2009, the first oral factor IIa (thrombin) inhibitor, dabigatran, was approved by the United States Food and Drug Administration (FDA) for the prevention of stroke in the setting of non-valvular atrial fibrillation (NVAF). Two oral factor Xa (fXa) inhibitors, rivaroxaban and apixaban, were subsequently FDA-approved for the same indication (Table 1) [8–11]. Eventually, these agents were all approved for the prevention and treatment of VTE as well. These non-vitamin K oral anticoagulants (NOACs) have challenged the previously accepted realities of therapeutic monitoring, bridging regimens, and dietary modifications. However, these agents are not without their clinical limitations. A number of pharmacokinetic drug interactions still must be considered for appropriate use and dosing. Additionally, apixaban and dabigatran require twice-daily dosing, which may negatively impact patient adherence. These NOACs have been found to be at least as safe as warfarin. While approved doses of rivaroxaban and dabigatran have resulted in similar rates of major bleeds compared to VKAs, apixaban is associated with a reduced risk [12]. The NOACs have resulted in a significant reduction in intracranial hemorrhage compared to VKAs, however several of these agents have been associated with an increase in gastrointestinal bleeding [13]. With these limitations in mind, the search for an optimal oral anticoagulant continues. Edoxaban (Savaysa), a new fXa inhibitor, was FDA-approved in 2015 and appears poised to address some of these practical concerns.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Edoxaban
Pharmacology
Edoxaban, like rivaroxaban and apixaban, is a selective fXa inhibitor. Edoxaban inhibits free fXa without the need of antithrombin (Fig. 1). This inhibition of fXa in the coagulation cascade leads to decreased thrombin generation, and therefore, a reduction in thrombus formation and progression. The reduction in thrombin also results in an indirect inhibition of platelet aggregation. Changes in the prothrombin time (PT), international normalized ratio (INR), and activated partial thromboplastin time (aPTT) may be observed in patients on therapeutic edoxaban doses. However, these changes tend to be small, unpredictable, and highly variable, so clinicians should not use these markers to monitor the anticoagulant effects or titrate the dose of edoxaban [14].

In a, FXa forms a complex with FVa to allow for conversion of prothrombin to thrombin. In b, FXa inhibitor binds to FXa, preventing the conversion of prothrombin to thrombin. In c, FXa inhibitor binds to andexanet alfa rather than FXa, allowing for formation of thrombin. Gla Gamma-carboxyglutamic acid
Pharmacokinetics
The pharmacokinetics of edoxaban have been shown to be dose dependent following single doses up to 150 mg and repeated doses up to 120 mg in healthy subjects [15, 16]. Peak plasma concentrations of the drug occur 1–2 h following oral administration. The oral bioavailability is approximately 62%, and unlike rivaroxaban, the systemic exposure of edoxaban does not seem to be affected by food. In vitro, edoxaban is approximately 55% protein bound. Unchanged edoxaban is the primary form in plasma. However, minimal metabolism via hydrolysis, conjugation, and oxidation by CYP3A4 is present [11]. In a study examining the elimination of radiolabeled edoxaban in healthy subjects, approximately 60% of the drug was eliminated in the feces, with approximately 35% eliminated in the urine. Over 70% of the drug was eliminated unchanged with an elimination half-life of 10–14 h [15, 16]. Patients with mild to moderate hepatic impairment do not appear to have any major changes in pharmacokinetic profiles. However, the pharmacokinetic profile is affected by renal impairment. Total systemic exposure increased 32% in patients with creatinine clearance (CrCl) 51–80 ml/min and 72% in patients with CrCl less than 30 ml/min, compared to patients with CrCl greater than 80 ml/min [11]. The clinical implications of these differences in pharmacokinetics will be discussed later.
Reversal
Similar to other NOACs, the need exists for a rapid reversal agent. Unlike the anticoagulant effects of warfarin, which can be reversed directly with the use of vitamin K, there is no approved reversal agent specific for the anticoagulant effects of edoxaban. Non-specific agents for reversal include prothrombin complex concentrate (PCC) and recombinant FVIIa. In a phase I trial evaluating PCC for the reversal of edoxaban in 110 subjects, PCC was found to reverse the anticoagulant effects of edoxaban 60 mg in a dose-dependent fashion. Complete reversal of bleeding duration and endogenous thrombin potential and partial reversal of PT was observed following a dose of 50 IU/kg [17]. While dialysis is an option for removal of dabigatran in the setting of acute bleeding due to its low percentage of protein binding and high renal clearance, it is not an effective option for reversal of fXa inhibitors. Edoxaban has a lower percentage of protein binding than the other fXa inhibitors, but its degree of renal clearance is much lower than that of dabigatran (Table 2) [8–11, 18]. In a study evaluating the pharmacokinetics and safety of edoxaban in patients undergoing hemodialysis, patients receiving hemodialysis (AUC0→∞ 676.2 ng h/ml) had only a minor decrease in mean total exposure compared to those patients off-dialysis (AUC0→∞ 691.7 ng h/ml), suggesting that hemodialysis would not be an efficient means of drug removal [19].
Several promising antidotes are currently in development for reversal of edoxaban and other NOACs. Andexanet alfa (PRT064445) is a modified, recombinant activated Factor X (rFXa) produced in Chinese hamster ovary cells. This modified rFXa lacks intrinsic procoagulant properties due to a mutation in the serine residue of the amino acid structure. To prevent competition between this inactive rFXa and active fXa molecules in the coagulation cascade, andexanet alfa lacks a membrane-binding γ-carboxyglutamic acid domain (Fig. 1). Therefore, andexanet alfa has the capacity to bind direct fXa inhibitors without interfering with the coagulation cascade [20, 21]. Several phase 1 and phase 2 trials have shown promise for andexanet alfa’s ability to reverse the effects of various fXa inhibitors [20]. One study specific to edoxaban showed that a single 60-mg dose of edoxaban was reversed 52% following a 600 mg bolus of andexanet and by 73% following an 800-mg bolus. Both bolus doses were followed by a continuous andexanet infusion of 8 mg/min for 1 h. Within 2 h of stopping the andexanet infusion, anti-fXa levels returned to those measured in the group receiving placebo infusions [22]. Phase 3 trials investigating this agent for reversal of NOACs are currently underway [23, 24].
Another promising antidote under development is aripazine (PER977). This small molecule binds non-covalently to and inhibits the activity of both direct and indirect anticoagulants, including both oral and parenteral agents. Unlike andexanet, which is specific to fXa inhibitors, aripazine has been shown to have effects on dabigatran as well [20]. Specific to edoxaban, a phase 1 trial was conducted in which 80 healthy volunteers received an intravenous bolus dose of 5–300 mg aripazine. In the patients who were also pretreated with 60 mg edoxaban, the whole blood clotting time was normalized following a single 300-mg bolus dose of aripazine. The anticoagulant effects of aripazine remained stable over a 24-h period [25]. An additional phase 2 trial investigating this agent for reversal of edoxaban is currently underway [26].
Drug Interactions
One benefit of the NOACs is the potential for fewer drug–drug interactions. However, several clinically significant drug interactions are worth noting with edoxaban. The majority of pharmacokinetic drug interactions result from inhibition or induction of the P-glycoprotein (P-gp) efflux transporter which is responsible for intestinal transport of edoxaban [27–30]. One study evaluated the effects of various cardiovascular agents on the pharmacokinetics of edoxaban (Table 3) [27–29]. Other drugs with pharmacokinetic interactions with edoxaban are worth noting. Ketoconazole, erythromycin, and cyclosporine also inhibit P-gp and concomitant use with edoxaban resulted in significant increases in Cmax and AUC. Rifampin, a P-gp inducer, when administered along with edoxaban resulted in significantly lower AUC [14]. Based on these interactions, patients taking protease inhibitors or cyclosporine were excluded from phase III clinical trials evaluating edoxaban [28, 29]. The Hokusai-VTE trial also required a dose reduction of edoxaban during any acute use of macrolide antibiotics, ketoconazole, or itraconazole. Chronic use of these antibiotics was not allowed [29]. The use of anticoagulants, antiplatelet drugs, and/or thrombolytics with edoxaban should be avoided due to the potential for increased risk of bleeding [11]. Patients receiving dual antiplatelet therapy were not enrolled in phase III clinical trials evaluating edoxaban [28, 29]. Low-dose aspirin, thienopyridines, and non-steroidal anti-inflammatory drugs were allowed in clinical trials evaluating edoxaban, and concomitant use of these medications resulted in increased rates of clinically relevant bleeding [11].
Atrial Fibrillation
Edoxaban was studied in a phase II trial for the prevention of stroke and systemic embolism in patients with NVAF [31]. This multicenter trial randomized 1146 patients to one of five treatment groups including edoxaban 30 mg PO daily (n = 235), 30 mg PO twice daily (n = 244), 60 mg PO daily (n = 234), 60 mg PO twice daily (n = 180), or warfarin titrated to a goal INR 2–3 (n = 250). While the primary outcome was the occurrence of major and/or clinically relevant non-major bleeding, the rate of stroke and systemic embolism was collected. Approximately 63% of patients had a CHADS2 score of 2 in each arm. The presence of stroke, TIA, or systemic embolism was low and no statistical differences were observed (0.4% edoxaban 30 mg daily; 1.3% edoxaban 30 mg twice daily; 0.4% edoxaban 60 mg daily; 1.1% edoxaban 60 mg twice daily; 1.6% warfarin). These data helped investigators chose the edoxaban 30- and 60-mg doses for the phase III investigation.
The ENGAGE AF-TIMI 48 trial was a double-blind, double-dummy, non-inferiority trial that randomized 21,105 patients to receive high-dose edoxaban (60 mg daily), low-dose edoxaban (30 mg daily), or warfarin titrated to a goal INR 2-3 [28]. Subjects were enrolled with documented NVAF with a CrCl greater than 30 ml/min. The mean CHADS2 score was 2.8 in each arm representing a moderate-to-severe risk population for the occurrence of the composite primary outcome, including stroke and non-CNS systemic embolism. The primary outcome occurred in 1.5% of patients receiving warfarin (1.18% high-dose edoxaban [Hazard Ratio (HR) 0.79; 97.5% confidence interval (CI) 0.63–0.99; p < 0.001] and 1.61% low-dose edoxaban [HR 1.07; 97.5% CI 0.87–1.31; p = 0.005]). The intention-to-treat analysis demonstrated a favorable trend with high-dose edoxaban (1.57% high-dose edoxaban versus 1.8% warfarin; p = 0.08) and an unfavorable trend with low-dose edoxaban (2.04% low-dose edoxaban; p = 0.10). In a pre-specified analysis, high-dose edoxaban significantly reduced the risk of the primary endpoint in patients who were warfarin-naive patients (HR 0.71; 95% CI 0.56–0.91] [32]. At the end of the study period, patients in each edoxaban arm were given half of the treatment dose until they achieved a therapeutic INR on warfarin. The purpose of this bridging approach was to reduce the risk of thrombotic complications during anticoagulant transition, as observed with rivaroxaban in the ROCKET-AF trial [33]. Patients randomized to the high-dose edoxaban were given edoxaban 30 mg daily and the low-dose edoxaban patients were given edoxaban 15 mg daily. Stroke and systemic embolism were evenly distributed across the three treatment groups (seven in each group). The impact of CrCl on the occurrence of stroke and systemic embolism was also closely monitored. The prevention of thrombosis appeared to diminish in patients with a higher CrCl. The potential benefits of edoxaban 60 mg compared to warfarin were absent with a CrCl of 95 ml/min or greater (HR 1.02 95% CI 0.76–1.38). This directly led to the FDA approved recommendations to avoid edoxaban in NVAF with a CrCl greater than 95 ml/min [34].
The safety of antithrombotic therapy with edoxaban in NVAF has been evaluated in multiple phase II clinical trials. The major phase II study conducted by Weitz et al. described the two once-daily edoxaban regimens (30 mg once daily and 60 mg once daily) to have similar safety profiles to warfarin, with less bleeding than the twice-daily regimens [31]. Pharmacokinetic analysis demonstrated that the risk of bleeding correlated best with trough levels, which were lower with the once-daily regimens, thus yielding a lower bleeding rate. Additional phase II trials added to the data reported by Weitz et al. by determining that dose adjustment of edoxaban may be required in patients with low body weight (≤60 kg) due to a higher incidence of all bleeding [35]. Pooled analysis of phase I and II trials also suggested that renal insufficiency (defined as CrCl 30–50 ml/min) and concomitant P-gp inhibitor treatment may also influence bleeding risk [36]. Concomitant aspirin administration may also be an important covariate for bleeding events, but this finding has not been shown to be statistically significant [35]. Overall, in phase II trials, edoxaban 30 and 60 mg once daily had rates of bleeding similar to or less than warfarin, produced treatment emergent adverse events similar to warfarin, and produced no significant difference in the incidence of hepatic enzyme elevations [31, 37]. From these trials, and results from pooled analyses, the once-daily edoxaban doses (30 and 60 mg) were selected for further investigation and comparison with warfarin for stroke prevention in patients with NVAF in the phase III ENGAGE AF-TIMI 48 trial [38]. Additionally, an edoxaban dose reduction (half dose) was recommended in patients with moderate renal impairment (CrCl 30–50 ml/min), body weight ≤60 kg, and in patients taking strong P-gp inhibitors [36].
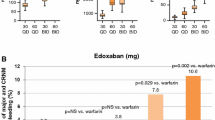
In the ENGAGE AF-TIMI 48 trial, the principal safety endpoint was adjudicated major bleeding (defined by the International Society on Thrombosis and Haemostasis) [28]. Other pertinent safety endpoints included any bleeding that was fatal, life-threatening, intracranial, or gastrointestinal, as well as any clinically relevant non-major bleeding (CRNMB), or the combination of any major or CRNMB. Both doses of edoxaban were superior to warfarin with respect to all significant bleeding outcomes including major, life-threatening, CRNMB, minor, and the combination of major or CRNMB. The annualized rates of major bleeding were 3.43% with warfarin, 2.75% with high-dose edoxaban (HR 0.80; 95% CI 0.71–0.91; p < 0.001), and 1.61% (HR 0.47; 95% CI 0.41–0.55; p < 0.001) with low-dose edoxaban (Table 4) [28]. With regards to the rate of gastrointestinal bleeding, low-dose edoxaban maintained superiority over warfarin (HR 0.67; 95% CI 0.53–0.83; p < 0.001), however high-dose edoxaban was associated with higher rates of gastrointestinal bleeding compared to warfarin (HR 1.23; 95% CI 1.02–1.50; p = 0.03). The rates of non-bleeding adverse events were similar in the three treatment groups. Overall, edoxaban (both high and low-dose) was shown to be superior to dose-adjusted warfarin in almost all major bleeding outcomes, and this superiority was maintained in the subgroup of patients with a CrCl <95 ml/min, as well as those who received a dosage reduction at randomization.
In an indirect treatment comparison analysis of the NOACs for stroke prevention in NVAF, some differential safety effects were discovered with edoxaban (both high- and low-dose) and the other agents [39]. Overall, high-dose edoxaban appears to have similar safety to other newer options but may be associated with lower risk of major (HR 1.30; 95% CI 1.08–1.57 with rivaroxaban) and major or CRNMB (HR 1.20; 95% CI 1.08–1.32 with rivaroxaban) compared to rivaroxaban and possibly increased risk of major or CRNMB compared with apixaban (HR 0.79; 95% CI 0.70–0.90 with apixaban). Low-dose edoxaban appears safer than all other oral agents in terms of bleeding events. These finding should be interpreted cautiously as there were important differences between the trials of these agents that could have led to the observations. Patient selection, quality of anticoagulation control with warfarin (reflected by average time in therapeutic range), and the presence of dosage reductions for particular patient characteristics were not consistent between trials. Additionally, although adjusted indirect treatment comparison meta-analysis is an established statistical technique, the data are generated using indirect evidence and therefore are not as precise as what would be possible with direct comparative data.
Venous Thromboembolism Prophylaxis
Edoxaban has not been FDA-approved for prophylaxis following orthopedic surgery. Most clinical studies have used comparators that are known to be inferior, unavailable in the United States, or never studied in phase III trials. The dose finding study for orthopedic surgery evaluated edoxaban 5 mg (n = 88), 15 mg (n = 92), 30 mg (n = 88), and 60 mg (n = 88) [40]. This multicenter, placebo-controlled trial randomized 523 patients following total knee arthroplasty. The occurrence of any thrombotic event (PE and DVT) was reduced with higher doses (29.5, 26.1, 12.5, 9.1%) versus placebo arm (48.3%). This dose-ranging trial helped establish the edoxaban doses to pursue in future phase III orthopedic prophylaxis trials. Three Japanese phase III trials were conducted with edoxaban for the prevention of VTE in the STARS (Studying Thrombosis After Replacement) program including the STARS E-3 trial (n = 716) following total knee arthroplasty, the STARS J-4 (n = 92) following hip fracture surgery, and the STARS J-V (n = 610) following hip arthroplasty [41–43]. The STARS E-3 trial was a double-blind, double-dummy trial that compared edoxaban 30 mg PO daily to enoxaparin 20 mg twice daily in patients following orthopedic surgery [41]. Patients undergoing total knee arthroplasty were randomized to receive edoxaban 30 mg daily (n = 360) 6–24 h post-surgery or enoxaparin 20 mg (n = 356) 24–36 h post-surgery. The primary outcome of this trial included symptomatic PE, and symptomatic or asymptomatic DVT. Edoxaban provided a statistically significant benefit compared to low-dose enoxaparin (7.9 vs. 13.9%; p = 0.010). No statistical differences in the individual outcomes of PE, symptomatic DVT, or asymptomatic DVT were observed. The STARS J-4 trial was a randomized, open-label trial that compared edoxaban 30 mg PO daily to enoxaparin 20 mg twice daily in patients undergoing hip fracture surgery [42]. Patients were randomized to receive edoxaban (n = 62) 6–24 h post-surgery or enoxaparin (n = 30) 24–36 h post-surgery for 11–14 days following hip fracture surgery. The secondary outcomes included efficacy endpoints including the incidence of thromboembolic events, venous thromboembolism-related deaths, and all-cause deaths. The incidence of thromboembolic events was 6.5% with edoxaban and 3.7% with enoxaparin, all of which were asymptomatic DVTs. The STARS J-V trial was a randomized, double-blind, non-inferiority trial that compared edoxaban and enoxaparin in Japanese patients undergoing total hip arthroplasty [43]. Patients were randomized to edoxaban 30 mg PO daily 6–24 h post-surgery or enoxaparin 20 mg twice daily 24–36 h post-surgery for 11–14 days. The primary efficacy outcome was defined as a composite outcome including symptomatic and asymptomatic DVT, and PE. The composite outcome occurred in 2.4% of edoxaban patients and 6.9% of enoxaparin patients (p < 0.001 for non-inferiority, p = 0.0157 for superiority). The difference in the primary outcome was largely driven by the incidence of asymptomatic DVT (2.4% edoxaban versus 6.5% enoxaparin). Fuji and colleagues studied the impact of renal function in patients treated with edoxaban following total knee arthroplasty, total hip arthroplasty, and hip fracture surgery [44]. Patients with CrCl 50–80 ml/min were given edoxaban 30 mg PO daily (n = 30), patients with CrCl between 20 and 30 ml/min were given edoxaban 15 mg PO daily (n = 22) or fondaparinux 1.5 mg SQ daily (n = 21), and patients with a CrCl between 15 and 20 ml/min were given edoxaban 15 mg PO daily. No significant reductions in embolism or death occurred in this population.
The safety of edoxaban for the prevention of VTE following orthopedic surgery was also assessed. In a Japanese patient population following total knee arthroplasty, edoxaban at doses of 5, 15, 30, and 60 mg once daily for 11–14 days produced no difference in major or CRNMB versus placebo (1.9–4.7% in edoxaban groups versus 3.9% in placebo group) [40]. The incidence of treatment-related all bleeding events (major, CRNMB, and minor) was significantly increased in the edoxaban 60 mg once-daily group compared with placebo (17 vs. 6.9%, respectively; p = 0.025), but was not increased with the other edoxaban doses (5.8, 10.4, and 10.7% with edoxaban 5, 15, and 30 mg once daily, respectively) [40]. Edoxaban at doses of 15, 30, 60, or 90 mg once daily for 7–10 days produced no significant difference in major and/or CRNMB versus dalteparin sodium (initially 2500 IU, followed by 5000 IU) once daily in a Caucasian population following total hip replacement, and had a comparable incidence of major and CRNMB versus subcutaneous enoxaparin sodium 20 mg twice daily following total hip arthroplasty [45]. Treatment emergent adverse events with edoxaban were similar to the comparator groups in all trials and there was no indication of hepatotoxicity. Overall, in phase II studies, edoxaban 30 mg once daily appeared to be well tolerated in both total knee and total hip arthroplasty and led to that dose being utilized for phase III testing.
Regarding safety, STARS J-4, J-V, and E-3 compared edoxaban 30 mg once daily to enoxaparin 20 mg twice daily (standard dosing in Japan), no significant difference in major and CRNM bleeding events was seen (major and CRNMB occurred in 6.2, 3.4, and 2.6% in the edoxaban groups in the STARS E-3, J-4 and J-V trials, respectively) [41–43]. However, these studies were generally underpowered to detect small differences in bleeding rates and the dosing of enoxaparin 20 mg twice daily may not allow extrapolation of these studies to other regions. Elevations in serum aminotransferase levels had a low incidence within the edoxaban groups and occurred less often than within the enoxaparin groups, a finding consistent with the results of a retrospective comparison between edoxaban, fondaparinux, and enoxaparin [46].
Venous Thromboembolism Treatment
The Hokusai-VTE trial was a double-blind, non-inferiority trial that compared the safety and efficacy of edoxaban in the treatment of VTE [29]. Investigators randomized 8292 patients to receive edoxaban 30 or 60 mg (n = 4118) or warfarin (n = 4122). All patients received open-label enoxaparin or unfractionated heparin for at least 5 days, with a median duration of 7 days. Edoxaban 60 mg (n = 3385) was recommended for patients with a CrCl greater than 50 ml/min, and edoxaban 30 mg (n = 733) was administered to patients with a CrCl between 30 and 50 ml/min. Warfarin was monitored and titrated to a goal INR 2–3 for 3–12 months, with 40% of patients receiving the full 12 months. Patients were considered for inclusion who experienced a DVT (n = 4921) and/or PE (n = 3319). The severity of the index event was similar in the two treatment groups. The recurrence of thromboembolism or VTE-related death occurred in 3.2% of edoxaban patients and 3.5% of warfarin patients (HR 0.89 95% CI 0.70–1.13; p < 0.001 for non-inferiority). No differences were observed with DVT alone, non-fatal PE, or fatal PE. Additionally, no differences in the primary outcome were seen in patients when the index event was a DVT or PE. The non-inferiority of edoxaban was maintained in those patients qualifying for the low dose compared to warfarin (HR 0.73 95% CI 0.42–1.26). Other trials examining target specific agents in the treatment of VTE have had a relatively small subgroup of patients with cancer at baseline. This trial has one of the largest cancer subgroups with 378 patients in the edoxaban arm (9.2%) and 393 patients in the warfarin arm (9.5%).
The primary safety outcome measured was the incidence of major or CRNMB [29]. Major bleeding was defined by the International Society on Thrombosis and Haemostasis and CRNMB was defined as any overt bleeding that did not meet the criteria for major bleeding but was associated with the need for medical intervention, contact with a physician, or interruption of the study drug or with discomfort or impairment of activities of daily life. The primary safety outcome occurred significantly less in the edoxaban group compared to warfarin, 8.5 and 10.3%, respectively (HR 0.81; 95% CI 0.71–0.94; p = 0.004) (Table 5) [29]. There were numerically fewer fatal and intracranial bleeding events with edoxaban; however, this was not statistically significant. Major bleeding within this patient group was also numerically lower with the reduced edoxaban dose (1.5% with edoxaban compared with 3.1% with warfarin, HR 0.50; 95% CI 0.24–1.03), but this was not statistically significant. Overall, the Hokusai-VTE trial established that in patients with acute VTE, edoxaban once daily was superior to warfarin with respect to bleeding, and in patients at a potential higher risk of bleeding (due to either renal impairment or low body weight), halving the dose of edoxaban to 30 mg significantly reduced bleeding while maintaining efficacy.
In an indirect treatment comparison analysis of the NOACs for the treatment of acute VTE, the adjusted risk of major bleeding was not significantly different between edoxaban and either rivaroxaban or dabigatran but did appear to be potentially increased in comparison to apixaban (Relative Risk 2.74; 1.40–5.39) [47]. The required use of parenteral anticoagulation treatment in the Hokusai-VTE trial, as well as a longer overall mean treatment duration (8.3 months versus 6 months, in Hokusai-VTE and AMPLIFY, respectively) could have potentially favored the lower bleeding with apixaban, so this finding should be interpreted cautiously. Additionally, though adjusted indirect treatment comparison meta-analysis is an established statistical technique, the data are generated using indirect evidence and therefore are not as precise as what would be possible with direct comparative data.
Clinical Considerations
Laboratory Monitoring
One of the biggest advantages of the NOACs is their predictable pharmacokinetic profile. This translates to the absence of routine monitoring in clinical practice. However, some clinical situations require the need for urgent and reliable monitoring such as surgery, overdoses, and bleeding. Edoxaban transiently increases the PT and aPTT in a concentration dependent manner; however, clotting times can significantly vary based on reagents and instrumentation. Furthermore, these coagulation assays measure the time to the initiation of clot formation and are not ideal to assess the effectiveness of edoxaban. One comparison measured the effect of edoxaban on seven PT reagents [48]. While the PT results linearly correlated to edoxaban plasma concentrations, the magnitude of the PT change among the reagents varied considerably. This variability persisted even when the PT was converted to an INR. While aPTT prolongation also increased with edoxaban in a concentration-dependent manner, the variability among eight aPTT reagents was smaller. While these assays might be helpful in evaluating edoxaban compliance, they are likely not helpful in evaluating the degree of anticoagulation or need for titration. The anticoagulant effect of edoxaban has also been measured using a thrombin generation assay in both platelet-poor and platelet-rich plasma. Edoxaban delayed both the initiation and propagation phase of thrombin generation. The thrombin generation assay was three times more sensitive to edoxaban than the PT and aPTT suggesting it could be used to measure edoxaban activity at trough concentrations. The dilute Russell’s viper venom time (dRVVT) may be another possible alternative to estimate the anticoagulation activity produced by edoxaban [49]. One study reported the anticoagulation effects of each NOAC, including edoxaban, using four dRVVT reagents. Each assay was highly sensitive and increased in a concentration-dependent manner with edoxaban. While this method is commercially available and easy to perform, it may not be routinely accessible at hospitals and is an expensive alternative.
Transition
Several clinical scenarios may occur that require initiation, discontinuation, or transition between edoxaban and a different anticoagulant. Bridging therapy is not necessary when initiating edoxaban due to its rapid onset of action. Edoxaban should be discontinued 24 h prior to surgical or other invasive procedures [11]. Transitioning from one anticoagulant to another should balance the risk for bleeding and thrombosis. When switching from warfarin, edoxaban should be initiated when the INR is <2.5. The conversion of edoxaban to warfarin can be accomplished using two different strategies. A parenteral agent can be initiated when the next edoxaban dose is scheduled, followed by a traditional bridging approach to warfarin. Edoxaban can also be converted to warfarin using half the edoxaban dose until a stable INR > 2.0 is achieved [33]. An oral transition from edoxaban to warfarin was successfully utilized in the ENGAGE AF-TIMI 48 trial to reduce the risk of thrombosis following edoxaban discontinuation.
Education
Patients should be involved in the decision to use edoxaban for approved indications balancing patient-specific factors (e.g., cost, health literacy, adherence) and medical factors (e.g., renal function, concomitant medications). Similar to other anticoagulants, patients prescribed edoxaban should be advised on the signs and symptoms of bleeding. Additionally, the patient should be instructed to take edoxaban at the same time each day and the importance of adherence should be emphasized as the anticoagulant activity returns to baseline at 24 h. While the need for routine therapeutic monitoring is not necessary, patients prescribed edoxaban may still benefit from frequent follow up to reinforce adherence and assess patient satisfaction. Historically, practitioners have focused on renal impairment when evaluating anticoagulation dosing. Practitioners should be reminded that the appropriateness of edoxaban requires evaluation of both normal and impaired renal function in the setting of NVAF. Given the number of indications and diversity in strategies used for initiation, transition, and discontinuation of each NOAC, general practitioners may benefit from a closed formulary or clinical assistance protocols.
Conclusions
Edoxaban has emerged as an alternative to an ever-growing class of selective oral anticoagulants. It has been shown to be a safe and effective option for the prevention of stroke in the setting of NVAF and the treatment of VTE. Edoxaban has several clinical advantages including a once-daily regimen, the lack of need for bridging or routine therapeutic monitoring, and absence of food-drug interactions. Renal function and P-gp drug interactions will challenge its acceptance as an alternative to traditional and other novel anticoagulants.
References
Heit JA. The epidemiology of venous thromboembolism in the community. Arterioscler Thromb Vasc Biol. 2008;28(3):370–2.
White RH. The epidemiology of venous thromboembolism. Circulation. 2003;107:I-4-I-8.
Fuster Valentin. ACC/AHA/ESC 2006 Guidelines for the Management of Patients with Atrial Fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Revise the 2001 Guidelines for the Management of Patients With Atrial Fibrillation): developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation. 2006;114(7):e257–354.
Kearon C, Gent M, Hirsh J, Weitz J, Kovacs MJ, Anderson DR, et al. A comparison of three months of anticoagulation with extended anticoagulation for a first episode of idiopathic venous thromboembolism. N Engl J Med. 1999;340(12):901–7.
Hart RG, Pearce LA, Aguilar MI. Meta-analysis: antithrombotic therapy to prevent stroke in patients who have nonvalvular atrial fibrillation. Ann Intern Med. 2007;146(12):857–67.
Beyth RJ, Quinn LM, Landefeld CS. Prospective evaluation of an index for predicting the risk of major bleeding in outpatients treated with warfarin. Am J Med. 1998;105(2):91–9.
The Stoke Prevention in Atrial Fibrillation Investigators. Bleeding during antithrombotic therapy in patients with atrial fibrillation. Arch Intern Med. 1996;156:409–16.
Pradaxa (dabigatran) [prescribing information]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals, Inc; 2015.
Xarelto (rivaroxaban) [prescribing information]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2015.
Eliquis (apixaban) [prescribing information]. Princeton, NJ: Bristol-Myers Squibb Company and New York, NY: Pfizer Inc; 2015.
Savaysa (edoxaban) tablets [prescribing information]. Parisippany, NJ: Daiichi Sankyo, Inc; 2015.
Gonsalves WI, Pruthi RK, Patnaik MM. The new oral anticoagulants in clinical practice. Mayo Clin Proc. 2013;8(5):495–511.
Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomized trials. Lancet. 2014;383:955–62.
Plitt A, Giugliano RP. Edoxaban: review of Pharmacology and key phase I to III clinical trials. J Cardiovasc Pharmacol Ther. 2014;19(5):409–16.
Masumoto H, Yoshigae Y, Watanabe K, Takakusa H, Okazaki O, Izumi T. In vitro metabolism of edoxaban and the enzymes involved in the oxidative metabolism of edoxaban [abstract]. AAPS J. 2010;12(S2):W4308.
Bathala MS, Masumoto H, Oguma T, He L, Lowrie C, Mendell J. Pharmacokinetics, biotransformation, and mass balance of edoxaban, a selective, direct factor Xa inhibitor, in humans. Drug Metab Dispos. 2012;40:2250–5.
Zahir H, Brown KS, Vandell AG, et al. Edoxaban effects on bleeding following punch biopsy and reversal by a 4-factor prothrombin complex concentrate. Circulation. 2015;131:82–90.
Coumadin (warfarin sodium) tablets [prescribing information]. Princeton, NJ: Bristol-Meyers Squibb, Inc; 2011.
Parasrampuria DA, Marbury T, Matsushima N, et al. Pharmacokinetics, safety, and tolerability of edoxaban in end-stage renal disease subjects undergoing haemodialysis. Thromb Haemost. 2015;113:719–27.
Greinacher A, Thiele T, Selleng K. Reversal of anticoagulants: an overview of current developments. Thromb Haemost. 2015;113:931–42.
Genmin Lu, DeGuzman FR, Hollenbach SJ, et al. A specific antidote for reversal of anticoagulation by direct and indirect inhibitors of coagulation factor Xa. Nat Med. 2013;19(4):446–53.
Crowther MA, Levy G, Lu G, et al. A Phase 2 randomized, double-blind, placebo-controlled trial demonstrating reversal of edoxaban-induced anticoagulation in healthy subjects by andexanet alfa (PRT064445), a universal antidote for factor Xa inhibitors. ASH 2014, 6-9.12.2014; Abstract 4269.
Portola Pharmaceuticals. A study in older subject to evaluate the safety and ability of andexanet alfa to reverse the anticoagulation effect of rivaroxaban. In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2000 [cited 2015 Jul 9]. NLM Identifier: NCT02220725. https://clinicaltrials.gov/ct2/show/NCT02220725. Accessed 24 Feb 2016.
Portola Pharmaceuticals. A study in older subject to evaluate the safety and ability of andexanet alfa to reverse the anticoagulation effect of apixaban. In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2000 [cited 2015 Jul 9]. NLM Identifier: NCT02207725. https://clinicaltrials.gov/ct2/show/NCT02207725. Accessed 24 Feb 2016.
Ansell JE, Bakhru SH, Laulicht BE, et al. Use of PER977 to reverse the anticoagulant effect of edoxaban. N Engl J Med. 2014;371(22):2141–2.
Perosphere, Inc. Study of PER977 administered to subjects with steady state edoxaban dosing and re-anticoagulation with edoxaban. In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2000 [cited 2015 Jul 9]. NLM Identifier: NCT02207257. https://clinicaltrials.gov/ct2/show/NCT02207257. Accessed 24 Feb 2016.
Mendell J, Zahir H, Matsushima N, Noveck R, Lee F, Chen S, Zhang G, Shi M. Drug–drug interaction studies of cardiovascular drugs involving P-glycoprotein, and efflux transporter, on the pharmacokinetics of edoxaban, an oral factor Xa inhibitor. Am J Cardiovasc Drugs. 2013;13:331–42.
Giugliano RP, Ruff CT, Braunwald E, For the ENGAGE AF-TIMI 48 Investigators, et al. Edoxaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2013;369:2093–104.
Hokusai-VTE Investigators. Edoxaban versus warfarin for the treatment of symptomatic venous thromboembolism. N Engl J Med. 2013;369:1406–15.
Mikkaichi T, Yoshigae Y, Masumoto H, Imaoka T, Rozehnal V, Fischer T, et al. Edoxaban transport via P-glycoprotein is a key factor for the drug’s disposition. Drug Metab Dispos. 2014;42(4):520–8.
Weitz JI, Connolly SJ, Patel I, Salazar D, Rohatagi S, Mendell J, Kastrissios H, Jin J, Kunitada S. Randomised, parallel-group, multicentre, multinational phase 2 study comparing edoxaban, an oral factor Xa inhibitor, with warfarin for stroke prevention in patient with atrial fibrillation. Thromb Haemost. 2010;104:633–41.
O’Donoghue ML, Ruff CT, Giugliano RP, Murphy SA, Grip LT, Mercuri MF, et al. Edoxaban vs. warfarin in vitamin K antagonist experienced and naïve patients with atrial fibrillation. Eur Heart J. 2015;36:1470–7.
Ruff CT, Giugliano RP, Braunwald E, Mercuri M, Curt V, Betcher J, et al. Transition of patients from blinded study drug to open-label anticoagulation. The ENGAGE-AF TIMI 48 trial. JACC. 2014;64(6):576–84.
Daiichi Sankyo, Inc. SAVAYSA (edoxaban tosylate). FDA draft briefing document for the cardiovascular and Renal Drugs Advisory Committee. NDA 206316. Meeting date October 30, 2014. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/CardiovascularandRenalDrugsAdvisoryCommittee/UCM420704.pdf. Accessed 24 Feb 2016.
Yamashita T, Koretsune Y, Yasaka M, Inoue H, Kawai Y, Yamaguchi T, Uchiyama S, Matsumoto M, Ogawa S. Randomized, multicentre, warfarin-controlled phase II study of edoxaban in Japanese patients with non-valvular atrial fibrillation. Circ J. 2012;76:1840–7.
Salazar DE, Mendell J, Kastrissios H, Green M, Carrothers TJ, Song S, Patel I, Bocanegra TS, Antman EM, Giugliano RP, Kunitada S, Dornseif B, Shi M, Tachibana M, Zhou S, Rohatagi S. Modelling and simulation of edoxaban exposure and response relationships in patients with atrial fibrillation. Thromb Haemost. 2012;107(5):925–36.
Chung N, Jeon HK, Lien LM, Lai WT, Tse HF, Chung WS, Lee TH, Chen SA. Safety of edoxaban, an oral factor Xa inhibitor, in Asian patients with non-valvular atrial fibrillation. Thromb Haemost. 2011;105(3):535–44. doi:10.1160/TH10-07-0451 Epub 2010 Dec 6.
Ruff CT, Giugliano RP, Antman EM, et al. Evaluation of the novel factor Xa inhibitor edoxaban compared with warfarin in patients with atrial fibrillation: design and rationale for the effective anticoagulation with factor Xa next generation in atrial fibrillation-thrombolysis in myocardial infarction study 48 (ENGAGE AF-TIMI48). Am Heart J. 2010;160:635–41.
Skjøth F, Larsen TB, Rasmussen LH, Lip GY. Efficacy and safety of edoxaban in comparison with dabigatran, rivaroxaban and apixaban for stroke prevention in atrial fibrillation. An indirect comparison analysis. Thromb Haemost. 2014;111(5):981–9.
Fuji T, Fujita S, Tachibana S, Kawai Y. A dose-ranging study evaluating the oral factor Xa inhibitor edoxaban for the prevention of venous thromboembolism in patients undergoing total knee arthroplasty. J Thromb Haemost. 2010;8:2458–68.
Fuji T, Wang CJ, Fujita S, et al. Safety and efficacy of edoxaban, and oral factor Xa inhibitor, versus enoxaparin for thromboprophylaxis after total knee arthroplasty: The STARS E-3 Trial. Thromb Res. 2014;134:1198–204.
Fuji T, Fujita S, Kawai Y, Nakamura M, Kimura T, Kiuchi Y, Abe K, Tachibana S. Safety and efficacy of edoxaban in patients undergoing hip fracture surgery. Thromb Res. 2014;133:1016–22.
Fuji T, Fujita S, Tachibana S, et al. Efficacy and safety of edoxaban versus enoxaparin for the prevention of venous thromboembolism following total hip arthroplasty: STARS J-V trial. Blood. 2010;116:Abstract 3320.
Fuji T, Fujita S, Kawai Y, Abe Y, Kimura T, Fukuzawa M, Abe K, Tachibana S. A randomized, open-label trial of edoxaban in Japanese patients with severe renal impairment undergoing lower-limb orthopedic surgery. Thromb J. 2015;13(6):1–11.
Fuji T, Wang C-J, Fujita S, et al. Safety and efficacy of edoxaban, an oral Xa inhibitor, for thromboprophylaxis after total hip arthroplasty in Japan and Taiwan. J Arthroplast. 2014;29:2439–46.
Sasaki H, Ishida K, Shibanuma N, Tei K, Tateishi H, Toda A, Yamashiro Y, Matsumoto T, Kuroda R, Kurosaka M. Retrospective comparison of three thromboprophylaxis agents, edoxaban, fondaparinux, and enoxaparin, for preventing venous thromboembolism in total knee arthroplasty. Int Orthop. 2014;38:525–9.
Kang N, Sobieraj DM. Indirect treatment comparison of new oral anticoagulants for the treatment of acute venous thromboembolism. Thromb Res. 2014;133(6):1145–51.
Morishima Y, Kamisato C. Laboratory measurements of the oral direct factor Xa inhibitor edoxaban. Comparison of prothrombin time, activated partial thromboplastin time, and thrombin generation assay. Am J Clin Pathol. 2015;143:241–7.
Exner T, Ellwood L, Rubie J, Barancewicz A. Testing for new oral anticoagulants with LA-resistant Russells viper venom reagents. An in vitro study. Thrombo Haemost. 2013;109(4):1–4.
Acknowledgments
No funding or sponsorship was received for this study or publication of this article. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Disclosures
Zachary Stacy is a consultant for Janssen Pharmaceuticals and Daiichi-Sankyo. William B. Call, Aaron P. Hartmann, Golden L. Peters and Sara K. Richter have nothing to disclose.
Compliance with ethics guidelines
This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Stacy, Z.A., Call, W.B., Hartmann, A.P. et al. Edoxaban: A Comprehensive Review of the Pharmacology and Clinical Data for the Management of Atrial Fibrillation and Venous Thromboembolism. Cardiol Ther 5, 1–18 (2016). https://doi.org/10.1007/s40119-016-0058-2
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40119-016-0058-2