
Abstract
The article presents application of homo polymer of dodecyl methacrylate (DDMA) and its copolymers with vinyl acetate (VA) as multifunctional additives for lubricant formulation. Homo polymer of DDMA and five copolymers of DDMA with VA at different molar ratios were synthesized by free radical polymerization method using azobisisobutyronitrile (AIBN) as initiator. The characterization of the polymers was carried out through FTIR, NMR and GPC (gel permeable chromatography) analysis. The performance of all the polymers as viscosity index improver (VII) or viscosity modifier and pour point depressant (PPD) additive in two different base oils (mineral) were evaluated. The mechanism of action of the polymers as pour point depressant was studied by photo micrographic analysis. Rheological study of the formulated lubricant was also carried out and reported. The thermal stability of the polymers was determined by thermogravimetric analysis (TGA). It was found that thermal stability, VI and molecular weights of copolymers are higher than the homopolymer which showed better PPD property.
Similar content being viewed by others
Introduction
Lubricating oils play a very important role in automobile industry. It keeps the moving parts lubricated and protects them against rust and corrosion. Lubricating oils alone cannot satisfy all the requirements of modern engines. Some additives are to be blended with lube oil to improve the overall performance of the lubricant [1]. The role of additives in lubricant is very significant. They optimize the performance of lubricant and generally fall into two major categories viz. surface active additive and performance enhancing additive [2]. The first one protects the metal surfaces of the engine from corrosion, such as antiwear, anti-rust and extreme pressure additives [3]. The second type reinforces the base stock performance, such as viscosity index improver (VII) [4, 5], pour point depressant (PPD) [6, 7], antioxidant [8], and detergent–dispersant [9]. Generally, in the formulation of a high performance lubricant, different types of additives at different percentages are blended with the base stocks. This increases the overall cost of the furnished lubricant. The addition of additives having multifunctional character to the base fluid may lead to formulate a cost effective as well as better performing lubricant. Therefore, research in this area has attracted much attention. In this work we have synthesized multifunctional additives which showed excellent VII and PPD performances. Lot of works in this direction has carried out so far. Kamal and his group [10] in their work have shown the application of copolymers of vinyl acetate (VA) and esters of acrylic acid as viscosity index improver for lubricating oil. They also studied the rheological properties of lube oil with and without polymeric additives. Al-Sabagh and his group [11] have mentioned the application of copolymers of VA with n-alkyl itaconate as pour point depressants in lubricating oil. The rheological properties were also studied by the same research team. Abdel-Azim et al. [12] has synthesized the copolymers of dialkyl fumarate with vinyl acetate and recognized that the copolymer of didodecyl fumarate with VA is the most effective as PPD for lubricating oil. In 2008, Nassar [13] had prepared six polymers at different molar ratios of 2-ethylhexyl methacrylate and vinyl acetate and studied the VI property of the lubricants. He reported that most efficient VI property is obtained when the ratio of acrylate and vinyl acetate is 1:0.2. The potential application of copolymer of vinyl acetate (VA) as pour point depressant for crude oil is also well documented. VA—α olefin copolymers [14] and VA—methacrylate copolymers [15] were used as pour point depressant for crude oil. The copolymers of vinyl acetate, styrene and n-butyl acrylate having different monomer ratios were used to study the rheological behaviours of Mexican crude oil [16]. Borthakur and his group [17] have shown the application of alkyl fumarate and vinyl acetate copolymer in combination with alkyl acrylate as a flow improver for high waxy Borholla crude oil. Machado et al. [18] studied the influence of ethylene vinyl acetate copolymers on viscosity and pour point of a Brazilian crude oil. Al-Shafy and Ismail prepared an ester of polyethylene acrylic acid with 1-docosanol and finally it was grafted with vinyl acetate to produce graft ester. The grafted product was used as flow improver for Egyptian waxy crude oils [19].
Although there exists lot of works on the additive performance of copolymers of VA and different acrylates, reports regarding their application as multifunctional additive like PPD and VII for lube oil are scanty. Therefore, in this work copolymers of DDMA with VA at different molar ratio were synthesized and their performances are evaluated. Here efficiencies of the polymers as viscosity index improver and pour point depressant in two different types of mineral base oils (SN150 and SN500) were carried out according to ASTM standards. Photo micrographic images were taken to study their mechanism of action as pour point depressant. Rheological properties of the lubricants were also studied during the work by a rheometer. Homo polymer of dodecylmethacrylate (DDMA) was also synthesized along with the copolymers for comparison of the results.
Experimental
Chemicals used
Methacrylic acid (MA, 99%, LOBA Cheme Pvt. Ltd.), vinyl acetate (VA, 99%, s.d fine chem Ltd.), dodecyl alcohol (DA, 98%, SRL Pvt. Ltd.) were used without purification. Toluene (99.5%, Merck Specialties Pvt. Ltd.), hexane (99.5%, s.d fine cheme Ltd.) and methanol (98%, Thomas Baker Pvt. Ltd.) were used after distillation. Hydroquinone (HQ, 99%, Merck Specialties Pvt. Ltd.) and azobisisobutyronitrile (AIBN, 98%, Spectrochem Pvt. Ltd.) were purified by recrystallization before use. Conc. H2SO4 (98%, Merck Specialties Pvt. Ltd.) was used as received. Base oils were collected from IOCL and BPCL, India and physical properties of them are given in Table 1.
Preparation of ester and its purification
Dodecylmethacrylate (DDMA) was prepared by reacting methacrylic acid with dodecyl alcohol in 1.1:1 molar ratio in presence of conc. H2SO4 as catalyst, 0.25% (w/w) hydroquinone (with respect to the total amount of the reactants) as polymerization inhibitor and toluene as solvent in a Dean Stark apparatus. The process of esterification was carried out by the procedure as reported in the earlier publication [20]. The purification of the ester was carried out by adding a suitable amount of charcoal to the prepared ester and refluxed for 3 h and then filtered off. The filtrate was treated with dilute (0.5 N) NaOH solution in a separating funnel to remove the unreacted acid and hydroquinone. The process was repeated several times and finally washed with distilled water. The purified ester was passed through sodium sulphate and left overnight over calcium chloride to dry completely and used in the polymerisation process.
Preparation of copolymers and homopolymer
The monomers, VA and DDMA, at different molar ratios (Table 2) were subjected to free radical polymerization using AIBN radical initiator. The polymerization was carried out in a three-necked round-bottom flask fitted with a magnetic stirrer, condenser, thermometer and an inlet for the introduction of nitrogen. In the flask, the mixture of DDMA and VA at a definite molar ratio in toluene was heated to 363 K for half an hour. AIBN (0.5% w/w with respect to the total monomer) was then added and heated for 6 h keeping the temperature constant at 363 K. At the end of the reaction time, the mixture was poured into cold methanol with stirring to terminate the polymerization and precipitate the polymer. The polymer was further purified by repeated precipitation of its hexane solution by methanol followed by drying under vacuum at 313 K. The homopolymer of DDMA was also prepared and purified in the same procedure. The synthesis of all the polymers is mentioned in Scheme 1.

Esterification, homopolymerization and copolymerization reaction
Measurements
Spectroscopic measurements
IR spectra were recorded by Shimadzu FTIR 8300 spectrometer using 0.1 mm KBr cell at room temperature within the wave number range of 400–4000 cm−1. NMR spectra were recorded in Bruker Avance 300 MHz FT-NMR spectrometer using a 5 mm BBO probe. CDCl3 was used as solvent and tetramethylsilane (TMS) as reference material.
Determination of the molecular weight
The number average molecular weight (M n) and weight average molecular weight (M w) were measured by GPC instrument (polystyrene calibration) equipped with a 2414 detector, waters 515 HPLC pump and 717 plus auto sampler. Sample solutions (0.4% w/v in HPLC grade THF) are prepared by dissolving ~4 mg of polymer per ml THF and filtering (0.45-μm Millipore PTFE) to remove suspended particulates. The pump flow rate is 1.0 mL/min with THF as the carrier solvent, and injection volumes are set to 20 μL. The polydispersity index [21] which indicates the nature of the distribution of the molecular weights in the polymers was also calculated.
Determination of thermo gravimetric analysis (TGA) data
The thermo-oxidative stability of all the polymers was determined by a thermo gravimetric analyzer (Shimadzu TGA-50) in air using an alumina crucible at a heating of rate of 10 °C/min. A comparison of thermal stabilities of homo polymer with the copolymers was explained by this study.
Performance evaluation as viscosity index improvers
Viscosity indices of the lubricating oils (SN150 and SN500) blended with polymers at different concentration levels [ranging from 1% to 5% (w/w)] were calculated by measuring kinematic viscosity (KV) values at 313 and 373 K. The ASTM D445 method was applied to determine the KV values and ASTM D 2270-10 method was applied to determine the VI values.
Performance evaluation as pour point depressants
Pour points were determined using the cloud and pour point tester (model WIL-471, India) according to ASTM D 97-09 method. The performance of additives as PPD was investigated through variation of their concentration [from 1 to 5% (w/w)] in the formulated lubricants.
Photographic analysis
The photomicrograph images showing wax behaviour of the lube oil (SN150, pour point = −6 °C) without and with polymers (4%, w/w) have been recorded at 0 °C. A Banbros polarizing microscope (model BPL-400B) was used for this purpose and the adopted magnification was 200X.
Rheological study
Rheological study of the homo polymer and copolymers at 5% (w/w) concentration in SN150 oil was performed using Brookfield rheometer (Model DV-III ultra). Dynamic viscosity (cp) and shear rate (s−1) were measured at two temperatures, 40°C and 100 °C.
Results and discussion
Spectroscopic analysis
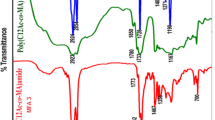
The homopolymer of DDMA exhibited IR absorption band at 1722.3 cm−1 for the ester carbonyl group. Peaks at 2853.5 and 2924 cm−1 were for the alkyl (CH3CH2–) groups. Peaks at 1456.2, 1435.9, 1377.3, 1330.2, 1296.1 and 1164.0 cm−1 were due to CO stretching vibration and absorption bands at 1013.5, 937.3, 812.9, 715.5, and 649.0 cm−1 were due to bending of C–H bonds.
In the 1H NMR of homopolymer, methyl protons appeared in the range of δ H 0.881–1.027 ppm, methylene protons in the range of 1.283–1.812 ppm for all alkyl groups. A broad peak at 3.930 ppm indicated the protons of –OCH2 group. Absence of any significant peaks in the range of 5–6 ppm in the spectrum confirmed completion of the polymerisation process. In the 13C NMR of homopolymer, peaks at δ C 176.73–177.84 ppm indicated the presence of ester carbons. The peaks at 63.06–65.40 ppm confirmed the presence of –OCH2 carbon. Peaks in the range of 14.13–45.22 ppm represent all sp3 carbon atoms of alkyl groups. No significant peaks in the range of 120–150 ppm indicated the absence of sp2 carbon atoms and therefore supported formation of the polymers.
The spectral data (IR and NMR) of all the five copolymers (P-2 to P-6) are similar. In the IR spectra, peaks at 1732.9–1735.2 and 1716.5–1720 cm−1 indicated the presence of ester carbonyl groups in the copolymers due to vinyl acetate and DDMA moiety, respectively. Peaks at 2853.6–2854 and 2922.9–2925.4 cm−1 were for the CH3CH2– groups. The peaks at 1456.2–1456.8 cm−1, 1377.1–1377.8 cm−1, 1368.4–1369 cm−1, 1321.1–1321.9 cm−1, 1296.1–1296.8 cm−1, 1238.2–1238.8 cm−1, 1163.0–1164.2 cm−1, 1065.5–1066 cm−1, and 1011.6–1012 cm−1 were due to CO stretching vibration and absorption bands at 814.9–816 and 721.3–721.9 cm−1 were due to bending of C–H bond. It is observed from the IR data of five copolymers that with increasing the percentage of vinyl acetate moiety in the copolymers the peak intensity olefinic groups gradually decreases.
In 1H NMR of copolymers, a broad peak δ 3.926–4.158 ppm indicated the protons of –OCH2 and –OCH3 groups. The hydrogen attached to sp3 carbons appeared in the range of 0.858–2.637 ppm. Absence of any significant peaks in the range of 5–6 ppm indicated the disappearance of C=C bonds and confirmed formation of the copolymers.
In 13C NMR, peaks at δ C 176.60–176.70 ppm indicated the presence of ester carbonyl groups. The peaks appeared from 64.66 to 65.06 ppm indicated the presence of –COCH3 methyl carbons and –OCH2 carbons. The peaks ranging from 14.08 to 45.09 ppm represented all other sp3 carbons. No significant peaks in the range of 120–150 ppm indicated the absence of sp2 carbons and confirmed the formation of the copolymers. (All IR and NMR spectra are given in supporting information).
Analysis of molecular weight data
The experimental values of M n and M w of the homo and copolymers are given in Table 2. From the values, it is found that molecular weight of homopolymer is less than the copolymers. The molecular weights of copolymers increase with increasing the percentage of vinyl acetate moiety in the prepared copolymers. The comparatively higher PDI values of P-2 and P-3 indicated that their molecular weight distributions are wider and the copolymers are more branching compared to others. On the other hand, the lowest PDI value of P-6 indicated that the molecular weight distribution of the copolymer is narrow compared to others and the polymer is expected to be more linear which is also reflected of its highest thermal stability.
Analysis of thermo gravimetric data
From the TGA values (Fig. 1), it is found that homo polymer of DDMA (P-1) is thermally less stable than the copolymers. Generally, copolymers having vinyl acetate (VA) as a monomer has a head to tail structure. It may be due to fact that VA molecules add to the growing chain of the polymer through its –CH2– group and such type of addition involves less steric effect [22]. Polymers formed in this process always have a higher molecular weight and become thermally more stable [23]. In case of P-1, decomposition started at 150 °C with 12.5% weight loss and 75% weight was lost at 400 °C. Among the copolymers, decomposition of thermally most stable P-6 started at 220 °C with 10% weight loss and found 50% weight loss at 400 °C. It may be due to higher molecular weight and lower PDI value [24] of P-6. The order of thermal stability of the prepared polymers is P-1 < P-2 < P-3 < P-5 < P-4 < P- 6.

Thermal degradation of polymers
Analysis of viscosity index data
Viscosity index (VI) values of the lubricant compositions prepared by blending polymers at different concentration levels [1–5% (w/w)] with two different types of base oils were determined from the kinematic viscosity values of the blends measured at 40 °C and 100 °C. The experimental results are given in Fig. 2 (for SN150) and Fig. 3 (for SN500). It is found that VI values of all the polymers increase with increasing concentration of the additives in base oils. With increase in temperature, the lube oil viscosity decreases. But at higher temperature, due to expansion of the additive molecules, the size of the solvated additives increases. This increase in size of the solvated additives counterbalances the reduction of viscosity of the lubricants at higher temperature. Increase in concentration of polymers in lube oil also leads to increase of the total volume of solvated polymer molecules, thus exerting higher thickening effect and improves the VI property [25–27]. From the experimental values, it is found that the VI values of homo polymer (P-1) are less than copolymers. It may be due to low molecular weight of P-1 compared to others. The VI of copolymers increases with increasing the percentage of vinyl acetate which may be due to increase of molecular weight of the copolymers P-2 to P-6 [28]. The relation between molecular weights and VI values of the synthesized polymers are also compared with the copolymers of 2-ethylhexyl methacrylate and VA published previously [13] and mentioned in Table 3. It is found that VI values increase with increasing the molecular weight in the polymers. For higher molecular weight polymers, due to higher degree of polymerization, the polymer units will be larger, i.e., their hydrodynamic volumes will definitely be greater that may be the reason of their higher VI values.

Variation of viscosity index of the base oil (SN150) blended with additives at different concentrations

Variation of viscosity index of the base oil (SN500) blended with additives at different concentrations
Analysis of pour point data
Pour points of the lubricant compositions prepared blending the polymers at different concentrations ranging from 1% to 5% (w/w) with the base stocks were tested and experimental values are given in Figs. 4 and 5. From the values, it is observed that all the polymers can be used effectively as PPD and the efficiency increases with increasing concentration of the polymers in base oil up to 4%. This means that within this concentration range, at lower temperature, the polymers may interact with the paraffinic wax of the lube oil and change their crystal sizes [29]. The homo polymer P-1 has greater efficiency as PPD compared to the copolymers. It may be due to higher polarity of P-1 compared to others. The highly polar polymers are greatly adsorbed by the wax crystals present in lube oil, and therefore efficiency as PPD of the polymers improves [30]. Among the five copolymers, the polymer P-2, P-3 and P-5 have greater efficiency as PPD. The higher polarity of may be due to higher PDI values of these three copolymers [31]. A polymer having higher polydispersity index (PDI) is generally branched chain polymer [32]. The branched chain polymers, due to higher polarity, can easily prevent formation of three-dimensional crystal network structures of the wax particles at low temperature than the linear chain polymers of lower PDI values.

Variation of pour point of the base oil (SN150) blended with additives at different concentrations

Variation of pour point of the base oil (SN500) blended with additives at different concentrations
Analysis of rheological study
Rheological study was performed with lube oil (SN150) without and with 5% (w/w) polymers. The values of dynamic viscosity against shear rate at 40 °C and 100 °C are given in Figs. 6 and 7 respectively. The viscosity of the lube oil without any additives is approximately constant with increasing the shear rate. This indicates that pure lube oil behaves like a Newtonian fluid at any shear rate [33]. For lube oil with additives, it is found that at low shear rate (5–30 s−1), the viscosity gradually decreases and the lubricant behaves like a non- Newtonian fluid [34]. However, at high shear rate, viscosity of the lubricants approximately remains constant and behaves like Newtonian fluids [35]. At low temperature, viscosity modifiers in lube oil exist as spherical coil having random orientation and exert high viscosity in the absence of shear. When shear is applied, the additives start rearranging themselves in the direction of flow and viscosity of the lubricants decrease [36]. At high shear rate, all the polymers are arranged in the direction of flow and there is a negligible change in viscosity. At higher temperature, viscosity modifiers exist in expanded form (Fig. 8) and hence polymers are easily arranged in the direction of flow under shear and viscosity approximately remains constant under high shear rate for all polymer blended lube oil. In this way, the copolymers counterbalance decrement of lube oil viscosity under high temperature. The decrease of viscosity was caused due to applying of shear and changing temperature from 40 °C to 100 °C. The higher of dynamic viscosities of the polymers from P-6 to P-1 at any shear rate is may be due to incorporation of VA in the backbone of DDMA which results in their higher average molecular weights and therefore greater volume.

Variation of dynamic viscosity with shear rate at 40 °C

Variation of dynamic viscosity with shear rate at 100 °C

Effect of temperature on polymer in lube oil
Analysis of photo micrographic image
Photo micrographic analysis of the lube oil (SN150) without and with 4% (w/w) additives was carried out to study the effect of additives as PPD and its mechanism. The photograph images are shown in the Fig. 9a–g. In Fig. 9a, the photograph of lube oil (pour point = −6 °C) without any additives, there is a large number of cyclic crystalline and some needle shaped waxes. The Fig. 9b–g represented the base oil blended with 4% of P-1, 4% of P-2, 4% of P-3, 4% of P-4, 4% of P-5 and 4% of P-6, respectively. Greater wax modification is found when the lube oil is blended with P-1(homo polymer) and least wax modification is observed in case of P-6. This indicates that homo polymer of DDMA is better as PPD compared to its copolymers with VA. This is in agreement with the pour point values determined by ASTM D97-09 method. Length of the largest crystalline waxes in Fig. 9a–g are 447.3, 120.6, 192.2, 223, 242.5, 226.8 and 268.7 μm, respectively.

Photomicrograph images of a pure lube oil (SN150), b lube oil + 4% (w/w) of P-1, c lube oil + 4% (w/w) of P-2, d lube oil + 4% (w/w) of P-3, e lube oil + 4% (w/w) of P-4, f lube oil + 4% (w/w) of P-5, g lube oil + 4%(w/w) of P-6
Conclusion
From the above study it is found that polymers are effective as viscosity index improver and pour point depressant for lube oil. Viscosity index property of homo polymer is lower than copolymers. VI property increases with increase in the percentage of vinyl acetate in the copolymers. The efficiency as pour point depressant was found higher in case of homo polymer compared to copolymers. Among the copolymers, the polymers which have higher PDI values, results better PPD property. From rheological study, it was found that pure lube oil is a Newtonian fluid at any shear rate but polymer doped lube oil is non-Newtonian fluid at low shear and Newtonian fluid at high shear rate.
References
Ghosh P, Das T (2011) Copolymer of decyl acrylate and styrene: synthesis, characterization and viscometric studies in different base stock. Adv Appl Sci Res 2:272–283
Mortier RM, Orszulik SY (1997) Chemistry and technology of lubricants, 2nd edn. Chapman & Hill, London
Miwa TK, Rothfus JA, Dimitroff E (1979) Extreme-pressure lubricant tests on jojoba and sperm whale oils. J Am Oil Chem Soc 56:765–770
Akhmedov AI (1994) Copolymer of alkyl methacrylates with styrene as V. I. improvers for lubricating oils. Chem Tech Fuels Oil 30:34–37
Jerbic IS, Vukovic JP, Jukic A (2012) Production and application properties of dispersive viscosity index improvers. Ind Eng Chem Res 5:11914–11923
Jung KM, Chun BH, Park SH, Lee CH, Kim SH (2011) Synthesis of methacrylate copolymers and their effects as pour point depressants for lubricating oil. J Appl Polym Sci 120:2579–2586
Ghosh P, Das T, Nandi D, Karmakar G, Mandal A (2010) Synthesis and characterization of biodegradable polymer—used as a pour point depressant for lubricating oil. Int J Polym Mater 59:1008–1017
Nassar AM, Ahmed NS, Abd El-Aziz KI, Abdel-Azim AAA, El-Kafrawy AF (2006) Synthesis and evaluation of detergent/dispersant additives from polyisobutylene succinimides. Int J Polym Mater 55:703–713
Selezneva IE, Levin AY, Monin SV (1999) Detergents–Dispersants additives for motor oils, alkylphenolates. Chem Technol Fuels Oils 35:389–395
Abdir MF, Ashour FEl-Z, Ahmed NS, Kamal RS, El-Zahed SM (2014) The effect of some additives on the rheological properties of engine lubricating oil. Int J Eng Res Appl 4:169–183
Al-Sabagh AM, Khalil TM, Sabaa MW, Khidr TT, Saad GR (2012) Poly (n-alkyl itaconate-co-vinyl acetate) as a pour point depressant for lube oil in relation to rheological flow properties. J Dispers Sci Technol 33:1649–1660
Abdel-Azim AAA, Abdel-Aziem RM (2001) Polymeric additives for improving the flow properties and viscosity index of lubricating oils. J Polym Res 8:111–118
Nassar AM (2008) The behaviour of polymers as viscosity index improvers. Pet Sci Technol 26:514–522
Abou El-Naga HH, Abd El-Azim WM, Ahmed MM (1985) Polymeric additives for pour point depression of residual fuel oils. J Chem Technol Biotechnol 35:241–247
Pederson KS, Ronningsen HP (2003) Influence of wax inhibitors on wax appearance temperature, pour point and viscosity of waxy crude oil. Energy Fuels 17:321–328
Castro LV, Vazquez F (2008) Copolymers as flow improvers for Mexican crude oil. Energy Fuels 22:4006–4011
Borthakur A, Laskar NC, Mazumdar RK, Rao KV, Subramanyam B (1995) Synthesis and evaluation of alkyl fumarate—vinyl acetate copolymers in combination with alkyl acrylate as flow improvers for Borholla crude oil. J Chem Technol Biotechnol 62:75–80
Machado Andre LC, Lucas Elizabete F (2007) The influence of vinyl acetate content of the poly (ethylene-co-vinyl acetate) (EVA) additive on the viscosity and the pour point of a Brazilian crude oil. Pet Sci Technol 19:197–204
Al-Shafy HI, Ismail EA (2014) Studies on the influence of polymeric additives as flow improvers for waxy crude oil. IOSR J Eng 4:54–61
Ghosh P, Das M, Upadhyay M, Das T, Mandal A (2011) Synthesis and evaluation of acrylate polymers in lubricating oil. J Chem Eng Data 56:3752–3758
Rogosic M, Mencer HJ, Gomzi Z (1996) Polydispersity index and molecular weight distributions of polymers. Eur Polym J 32:1337–1344
Marvel CS, Riddle EH (1940) The structure of vinyl polymers. J Am Chem Soc 62:2666–2670
Grassie N, Melville HW (1949) The thermal degradation of polyvinyl compounds. Proc R Soc Lon A 199:1–13
Ghosh P, Karmakar G (2012) Synthesis and characterisation of Polymyristyl acrylate as a potential additive for lubricating oil. Am J Polym Sci 2:1–6
Tanveer S, Prasad R (2006) Enhancement of viscosity index of mineral base oils. Ind J Chem Technol 13:398–403
Abdel-Azim AAA, Malcolm BH (1984) Interaction parameters in ternary polystyrene solution at high temperature. Polymer 25:803–807
Abdel-Azim AAA, Malcolm BH (1983) Viscometric behaviour of polystyrene in tetralin/cyclohexane mixture. Polymer 24:1429–1433
Nassar AM, Ahmed NS, Kamal RS, Abdel-Azim AAA, El-Nagdy EI (2005) Preparation and evaluation of acrylate polymers as viscosity index improvers for lube oil. Pet Sci Technol 23:537–546
El-Gamal IM, Atta AM, Al- Sabbagh AM (1997) Polymeric structures as cold flow improvers for waxy residual fuel oil. Fuel 76:1471–1478
Nassar AM, Ahmed NS (2006) The behavior of α-olefins butyl acrylate copolymers as viscosity index improvers and pour point depressants for lube oil. Int J Polym Mater 55:947–955
Al-Sabagh AM, Sabaa MW, Saad GR, Khidr TT, Khalil TM (2012) Synthesis of polymeric additives based on itaconic acid and their evaluation as pour point depressants for lube oil in relation to rheological flow properties. Egypt J Pet 21:19–30
Chunxia H, Stephane C, Paula W-A, John MD (2012) Molecular structure of high melt strength polypropylene and its application to polymer design. Polymer 44:7181–7188
Hassaneau MHM, Bartz WJ, Abou El-Naga HH (1997) A study of the rheological behaviour of multigrade oil. Lubr Sci 10:43–58
Chou CC, Lee SH (2008) Rheological behaviour and tribological performance of a nanodiamond-dispersed lubricant. J Mater Proc Technol 201:542–547
Punit K, Khonsari MM (2009) On the role of lubricant rheology and piezo-viscus properties in line and point contact EHL. Tribol Int 42:1522–1530
Ahmed NS, Nassar AM, Nassar RM, Abdel Raouf ME, El-Kafrawy AF (2014) The rheological properties of lube oil with terpolymeric additives. Pet Sci Technol 32:2115–2122
Acknowledgements
Authors thank UGC, New Delhi for financial support. Thanks also to IOCL, India for supplying base oil.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ghosh, P., Hoque, M., Karmakar, G. et al. Dodecyl methacrylate and vinyl acetate copolymers as viscosity modifier and pour point depressant for lubricating oil. Int J Ind Chem 8, 197–205 (2017). https://doi.org/10.1007/s40090-017-0119-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40090-017-0119-y