
Abstract
Immunotherapy, based on increasing knowledge of the mechanisms of immune-mediated elimination of tumor cells, is a new approach to lung cancer therapy. This paper reviews clinical experience of two types of immunotherapy for lung cancer. In the first approach antigen-independent immunomodulatory therapy is used to target crucial immune checkpoints. This strategy includes use of ipilimumab—to block the interaction of cytotoxic T-lymphocyte antigen-4 with CD80 and/or CD86, thereby enhancing T-cell proliferation—and the fully human monoclonal antibodies BMS-936558 and BMS-936559—to target the programmed cell death protein 1 signaling pathway, thereby enhancing T-cell proliferation, cytokine secretion, and cytolysis of target cells. The second approach is antigen-specific cancer immunotherapy, including vaccines against melanoma-associated antigen 3, mucinous glycoprotein-1, transforming growth factor-beta 2, epidermal growth factor, and, less specifically, whole-tumor cell extracts. An overview of the clinical data from these strategies is presented, with discussion of questions and issues that remain unanswered.
Similar content being viewed by others


Introduction
Worldwide, lung cancer is the most commonly occurring cancer, with an estimated 1.6 million new cases reported in 2008 [1]. In the same year, it was also reported to be the leading cause of global cancer deaths, accounting for the loss of life of nearly 1.4 million people [1, 2]. In the United States, it is estimated that over 226,000 new cases of lung cancer were diagnosed in 2012 and approximately 160,000 people died of the disease [3]. Most cases of lung cancer are diagnosed at an advanced stage, and five-year survival is nearly 15.6 % in the United States [4]. In Europe, lung cancer was reported to be the third most common cancer in 2006, accounting for 12.1 % of all new cases [5]. In 2008, the crude incidence of lung cancer in Europe was 52.5/100,000 per year, and mortality was 148,000 per year [6].
Lung cancer is a heterogeneous disease that includes two main histologic subtypes: non-small-cell lung cancer (NSCLC) and small-cell lung cancer (SCLC). More than 85 % of all lung cancer cases are NSCLC, which includes two major types:
-
1.
non-squamous carcinoma (including adenocarcinoma, large-cell carcinoma, and other cell types); and
-
2.
squamous cell carcinoma [4].
Although new chemotherapy and molecule-targeting agents have been introduced in the past decade, outcomes for patients with NSCLC remain poor, with overall cure rates less than 20 % [7]. Thus, it is clear that novel treatment approaches are warranted. This article describes how the immune system is intricately linked to tumor development, and reviews the rationale and use of immunotherapy in the management of NSCLC.
Involvement of the immune system in tumor development
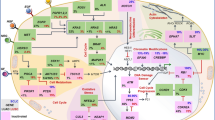
The immune system comprises two main branches: innate and adaptive immunity. Innate immunity relies on a set of germline-encoded pattern-recognition receptors that recognize components conserved among broad groups of microorganisms, or signals from damaged, injured, or stressed cells. The innate immune response is provided by granulocytes, macrophages, dendritic cells, natural killer (NK) cells, and complement. An adaptive response develops more slowly and comprises both a humoral and cell-mediated immune response, involving primarily B- and T-lymphocytes, respectively (Fig. 1). The humoral response is mediated by B-lymphocytes, which recognize “non-self” antigens, mature into plasma cells, then secrete specific antibodies. The antigen-presenting cells (APCs; commonly called dendritic cells) interact with naive T cells to trigger activation and proliferation of CD4+ helper and CD8+ cytotoxic T cells. This activation occurs after the interaction between the antigen major histocompatibility complex (MHC) on the APCs and T-cell receptors on the naive T cells, and a costimulatory interaction between CD80 or CD86 on the APCs and CD28 on the T cells [7]. During the cell-mediated immune response, phagocytes, NK cells, and antigen-specific cytotoxic T-lymphocytes are activated and a variety of cytokines are released in response to an antigen.

Schematic diagram of adaptive immunity [22]
Tumor cells can elicit an innate or adaptive immune response [8]. This response to tumor growth is commonly referred to as “cancer immunosurveillance”. Cancer immunosurveillance was first proposed in the early 1900s, but preclinical data supporting the idea that the immune system can actively eliminate tumor cells, and thereby prevent tumor growth, was only realized relatively recently by use of a variety of mouse models. After exposure to the carcinogen methylcholanthrene (MCA), lymphocyte-deficient, severe combined immunodeficient (SCID), and nude mice were all more susceptible to tumor induction [9–12]. Using at least three different mechanisms, the immune system helps to eliminate tumor cells [13] by:
-
1.
protecting the host from viral infection, thereby suppressing virus-induced tumors;
-
2.
preventing the development of an inflammatory environment—known to facilitate tumorigenesis [14, 15]; and
-
3.
eliminating tumor cells that express tumor-specific antigens, which are recognized by immune receptors on lymphocytes of the adaptive immune system.
The immune system not only acts to eliminate tumors, but also affects the immunogenicity of a given tumor. In an crucial experiment in which MCA-induced sarcomas from immunodeficient and immunocompetent mice were transplanted into naive, syngeneic wild-type mice, a significant proportion of those derived from immunodeficient mice were spontaneously rejected whereas all those induced in immunocompetent wild-type mice grew progressively [11]. This suggests that tumors formed in the absence of an intact immune system are more immunogenic than those developing in an immunocompetent host.
As a result of these discoveries, the cancer immunosurveillance hypothesis was further refined by proposing a dynamic interaction between the immune system and cancerous cells with three distinct phases and outcomes; this process has been termed cancer immunoediting [8, 13, 16]. The first phase of immunoediting consists of elimination, in which the innate and adaptive immune systems destroy the tumor before it becomes clinically apparent; this phase has never been directly observed, but its presence is inferred by comparing tumor development in wild-type mice with that in different types of immunodeficient mice [8]. In cases where tumor cells are not completely eliminated, they may enter the equilibrium phase, in which the immune system controls net tumor growth and the tumor cells are functionally dormant. Evidence for this phase of immunoediting comes from studies on immunocompetent mice, which have been shown to harbor latent cancer cells [17]. When the immune systems of the mice were ablated using monoclonal antibodies to deplete T cells and IFN-γ, tumors rapidly appeared at the original MCA injection site. Further analysis showed that the occult cancer cells were maintained in equilibrium by adaptive immunity (interleukin-12 [IL-12], IFN-γ, CD4+, and CD8+ T cells). In an additional scenario observed in a mouse model of melanoma, tumor cells disseminated throughout the body before the primary tumor was clinically detectable and remained dormant for different periods of time before metastatic growth occurred; in the lung, dormancy was mediated, at least in part, by cytostatic CD8+ T cells [18]. In the final escape phase, tumor cells emerge from functional dormancy and begin immunologically unrestricted growth. The different mechanisms involved can be broadly classified as either cell-autonomous modifications—at the level of the tumor cell—that enable cells to evade immune detection, or destruction and/or modification of immune cells effected by tumor cells, to generate an immunosuppressive network [8, 13]. Common changes to tumor cells include down-regulation of MHC expression, rendering them insensitive to T cells, and secretion of factors that suppress T cell responsiveness, for example immune-suppressive cytokines or enzymes able to catabolize amino acids critical for T cell effector function [19]. Tumors may also recruit or convert inflammatory cells that suppress T cell responses, including regulatory T cells (Treg), myeloid-derived suppressor cells (MDSC), and tolerogenic dendritic cells [20, 21].
Overall, involvement of the immune system is central to tumorigenesis. At best, the immune system suppresses tumor growth; at worst, the immune system promotes tumor progression either by selecting for tumor cells able to survive in an immunocompetent host or by establishing conditions within the tumor microenvironment that enable tumor metastasis.
Rationale for immunotherapy in NSCLC
Evidence of immunity in NSCLC
Increasing evidence suggests that many of the immune responses described above are present in lung tumors and, furthermore, that the extent of the immune response may correlate with patient outcome. Retrospective analysis has suggested that high tumor infiltration with CD4+ and/or CD8+ T cells is associated with improved survival of patients with early-stage (stages I to IIIA) NSCLC [23–25]. In another study, the presence of higher numbers of tumor-infiltrating CD8+ T cells in cancer nests compared with cancer stroma in patients with stage IV NSCLC was an independent predictor of better survival; similar results were found for infiltrating macrophages [26]. In support of this evidence, a mixed cohort study of patients with stages I to IV NSCLC found that the number of macrophages in the tumor islets or stroma was an independent predictor of survival time, and was more useful in predicting patient survival than counting mature dendritic or cytotoxic T cells [27].
There is also growing evidence of an immunosuppressive microenvironment in NSCLC tumors and its association with poor prognosis. Several studies have looked for the presence of CD4+ CD25+ Treg cells, known to reduce antitumor immunity, and their relationship with NSCLC stage and prognosis. Treg cells also constitutively express the transcription factor, Foxp3, and some studies have accordingly used this as a marker of Tregs in addition to (or instead of) assessing CD25 and CD4 expression. Higher levels of CD4+ CD25+ Treg cells have been observed in the peripheral blood of patients with NSCLC compared with healthy controls [28–31]. In some studies, the proportion of Treg cells (or, in one case, a subpopulation of Treg cells which also expressed CD13) was found to increase with NSCLC stage [31, 32], but decreased on surgical removal of tumors [32]. The proportion of Treg cells was also significantly higher in metastatic than non-metastatic stages [31], and another study found higher Foxp3 expression in metastatic lymph nodes than in non-metastatic nodes [30]. In a cohort of patients with stage I NSCLC, a higher ratio of Treg cells to tumor-infiltrating T cell lymphocytes was linked with significantly increased risk of disease recurrence [33]. Likewise, for patients with resected NSCLC, tumor-infiltrating Foxp3+ Tregs were associated with shorter recurrence-free survival [34]. In this study, Foxp3 expression was an independent predictor of significantly shorter recurrence-free survival in node-negative NSCLC, but not in the entire population of patients with NSCLC. A recent study confirmed that high Treg counts in surgical specimens from 87 patients with NSCLC were associated with worse overall survival (OS) and relapse-free survival [35]. Notably, another study failed to find any correlation between Foxp3-expressing tumor-infiltrating lymphocytes and clinicopathological features or prognosis in a cohort of 159 patients with stage III or IV NSCLC [36].
Tumor-associated antigens identified in NSCLC
Several tumor-associated antigens (TAA) known to be expressed in a range of different tumor types are also found in patients with NSCLC. The most commonly targeted antigens currently being investigated in clinical trials are listed briefly here. Mucinous glycoprotein-1 (MUC1) is a highly glycosylated transmembrane protein. In cancer cells, MUC1 often loses its polarity of expression, is overexpressed, or is under or aberrantly glycosylated, thereby exposing peptide epitopes [37]. In a cohort of patients with stage IB NSCLC, MUC1 was overexpressed in approximately 86 % of NSCLC adenocarcinomas, 39 % of squamous-cell tumors, and 74 % of other histology [38]. Furthermore, upregulated MUC1 expression was associated with poorer overall and disease-free survival in a cohort of patients with stage IB NSCLC, and was an independent prognostic factor [38].
Melanoma-associated antigen 3 (MAGE-A3) is a protein expressed almost exclusively on tumor cells. It is recognized by cytotoxic T cells in the presence of the human leukocyte antigen-A1 molecule. In NSCLC, MAGE-A3 expression has been reported in approximately 24–50 % of tumors [39–41], with expression more frequently occurring in histology of squamous cell, rather than adenocarcinoma [40–42]. Like MUC-1, expression of MAGE-A3 is associated with poor prognosis in NSCLC [43].
Gangliosides are a broad family of structurally-related glycolipids involved in cell–cell recognition, cell–matrix attachment, and differentiation. These glycolipids are believed to be good targets for cancer immunotherapy, because they are present at higher levels on tumor cells than on matched normal tissue. In the context of specific TAA, neu-glycosylated sialic acid-containing ganglioside (NeuGc-GM3) is a variant ganglioside identified almost exclusively in transformed cells [44], although little has been specifically published on its expression in NSCLC. Last, the epidermal growth factor receptor (EGFR) pathway, and specifically EGF itself, has been identified as a potential antigen to target in NSCLC.
Mechanistic rationale for immunotherapeutic techniques
Blockade of key immune checkpoints CTLA-4 and PD-1
The immune response to an antigen is regulated by a balance between co-stimulatory and inhibitory signals (or immune checkpoints) which, under normal conditions, are essential in maintaining self-tolerance and preventing autoimmunity, and in protecting normal tissue from damage when the immune system is responding to pathogenic infection. One key immune checkpoint that serves to limit activation of T cells by APCs is the cytotoxic T-lymphocyte antigen-4 (CTLA-4). Expression of CTLA-4 is upregulated on activation of the T-cell receptor complex [45]. CTLA-4 is a close homologue of CD28 (also expressed on the T-cell surface and, as discussed earlier, is responsible for the costimulatory interaction with APCs). Once expressed on the surface of T cells, CTLA-4 seems to directly compete with CD28 for CD80 and/or CD86 on the APCs, ultimately leading to T-cell inhibition (Fig. 2) [45–48].

Antagonistic action of CD28 and CTLA-4 on T cell activation: (a) co-stimulation via CD28 ligation transduces T-cell activating signals; (b) CTLA-4 ligation on activated T cells downregulates T-cell responses; and (c) blocking CTLA-4 ligation enhances T-cell responses. Adapted, with permission, from Shepherd F, Douillard J-Y, Blumenschein GR. Immunotherapy for non-small-cell lung cancer: novel approaches to improve patient outcome. J Thorac Oncol. 2011;6:1763–1773. APC, antigen-presenting cell; CTLA-4, cytotoxic T-lymphocyte antigen-4; MHC, major histocompatibility complex; TCR, T-cell receptor [7]
Preclinical studies have shown that in vitro inhibition of CTLA-4 by a monoclonal antibody (mAb) to prevent its interaction with CD80 and/or CD86, while preserving CD28 signaling, enhanced T cell proliferation, and secretion of IL-2 [46, 49]. In addition, injecting mice with an anti-CTLA-4 antibody resulted in expansion of CD8+ cells in response to superantigens [50]. Further evidence of the importance of CTLA-4 in inhibiting T-cell activation and proliferation comes from studies of CTLA-4-deficient mice. Extensive CD28-dependent expansion of autoreactive T cells is observed in these mice and they die within four weeks of birth after massive lymphoproliferation and lymphadenopathy [51, 52]. Taken together, these data identify CTLA-4 as a molecule involved in dampening T-cell activation, and have led to the hypothesis that inhibition of CTLA-4 could increase antitumor immunity mediated by effector CD4+ and CD8+ T cells.
Programmed cell death protein (PD-1) is a second type of T-cell surface receptor of crucial importance in checkpoint regulation during an immune response [53–55]. PD-1 is upregulated not only on activated T cells but also on activated B cells and several myeloid cells. PD-1 limits T-cell function, including proliferation, cytokine secretion, and cytolysis of target cells, by transduction of negative signals produced after binding to its ligands, PD-L1 and PD-L2. PD-L1 is expressed on many different types of tissue—both normal and tumoral. Once believed to be expressed on dendritic cells and monocytes only, PD-L2 expression has also been found on other cell types including normal and cancer-associated fibroblasts [56, 57]. In one study, both PD-L1 and PD-L2 were found to be expressed by tissue of patients with esophageal cancer, and patients who were PD-L1 or PD-L2-positive had a poorer prognosis than patients who were negative [58]. Both ligands are constitutively expressed in peripheral tissue during homeostasis and become elevated in response to tissue insult and inflammation [59]. The importance of PD-1 is apparent from PD-1-deficient mice, in which, after several weeks, spontaneous organ-specific autoimmunity is observed, depending on genetic background [60]. Moreover, PD-1-deficient mice have enhanced antitumor T-cell responses to a range of solid and hematopoietic tumors resulting in tumor regression and prolonged survival [61–63]. By comparison, tumors which overexpress PD-1 grow more aggressively in vivo [64]. Importantly, the phenotypic differences between PD-1 and CTLA-4 knockout mice suggest that each contributes uniquely to immune regulation. Similar to those for CTLA-4, these preclinical data suggest that PD-1 blockade may be an effective antitumor therapeutic strategy.
Modulation of dendritic cell phenotype with recombinant human lactoferrin
Changes in the phenotype and function of dendritic cells can also contribute toward immune tolerance of tumors. Fully mature dendritic cells can express a variety of T-cell costimulatory molecules able to induce immune reactions, many of which are lacking in non-activated dendritic cells. NSCLC has been shown to prevent induction of immunity by affecting dendritic cell phenotype and keeping these cells in an immature state [65, 66]. For example, B7-H3 is a member of a family of molecules known to inhibit immune reactions and to be upregulated in conditions promoting tumor growth [67]. Upregulation of this molecule in dendritic cells (as observed in NSCLC tumor samples) has been associated with impaired capacity to stimulate T-cell proliferation [68], although recently published data contradicts this finding in murine cancers and other human cancers [69].
Following these observations, strategies to encourage the immune system to recognize tumor antigens and shift the balance away from immune tolerance of tumors are being investigated for NSCLC. Lactoferrin, an immunomodulatory protein that enhances innate immunity and inflammation, has also been shown to affect the adaptive immune response by inducing maturation of dendritic cells. Talactoferrin, an orally active recombinant human lactoferrin that is being evaluated as an anticancer agent, interacts with gut epithelium and gut-associated lymphoid tissue, resulting in recruitment and maturation of immature dendritic cells. Although the actions of talactoferrin may occur at a location that is distant from primary NSCLC tumors, it is postulated (on the basis of preclinical data) that talactoferrin-induced maturation of dendritic cells in the presence of tumor antigens and immune effector cells may subsequently enhance immune priming with tumor antigens, thereby promoting tumor infiltration of CD8+ T cells and natural killer cells and resulting in immune-mediated tumor cell death [70, 71].
These strategies act on elements of the immune system in general; therefore, the above-described mechanisms of action are not dependent on the expression of any particular TAA. In contrast, research is revealing that specific TAAs may also be useful therapeutic targets, because of their common expression within this type of tumor and their persistence throughout disease progression, including on metastatic cells. For some, there is also evidence that the TAA promotes an immune response against the tumor.
Targeting tumor-associated antigens
An immune response to a tumor occurs when APCs internalize tumor cell fragments and then process and display the fragments on the cell surface in association with class I and II MHC molecules. As described above, these APCs interact with naive T cells to trigger activation and proliferation of tumor-specific CD4+ helper and CD8+ cytotoxic T cells, thereby turning an immunosuppressive environment into one that is immunostimulating—as with a preventive vaccine. The first step of active immunotherapy for NSCLC thus requires the ability of the host immune system to recognize the tumor as foreign, which depends on identification of antigens either exclusively or aberrantly expressed on the tumor cells (as described above). Inclusion of an adjuvant (a non-specific immune stimulant that promotes movement of APCs to the vaccination site and uptake of the vaccine by the APCs) in antigen-specific vaccines is believed to be essential.
Clinical experience with antigen-independent immunomodulatory therapy in NSCLC
Significant clinical experience has been gained with antigen-independent immunomodulatory therapy (Table 1). Checkpoint blockade by use of anti-CTLA-4 monoclonal antibodies, for example ipilimumab, is one of the most promising approaches. Ipilimumab is a fully human mAb that specifically blocks the binding of CTLA-4 to its CD80 and/or CD86 ligands, thereby augmenting T-cell activation and proliferation, and promoting tumor infiltration by T cells and tumor regression in animal models [47]. Subsequent early phase I and II studies revealed antitumor activity of ipilimumab across a range of tumor types. In two phase III trials on advanced melanoma, ipilimumab had a statistically significant OS benefit as monotherapy [72] or in combination with dacarbazine [73] either for previously treated or untreated patients. Treatment-related adverse events, for example diarrhea, pruritus, rash, and fatigue, were managed by following product-specific guidelines [72, 73].
The therapeutic potential of ipilimumab for patients with chemotherapy-naive advanced (stage IIIB or IV) NSCLC and extensive disease small-cell lung cancer (ED-SCLC) has been assessed in a phase II study [74•, 75]. Patients were randomized 1:1:1 to receive carboplatin (area under the curve, 6) and paclitaxel (175 mg m−2) combined with either placebo (up to six doses; control regimen), or ipilimumab 10 mg kg−1 on either a concurrent regimen (four doses of ipilimumab plus carboplatin and paclitaxel followed by two doses of placebo plus chemotherapy), or a phased regimen (two doses of placebo plus chemotherapy followed by four doses of ipilimumab plus chemotherapy). Eligible patients continued ipilimumab or placebo every 12 weeks as maintenance therapy. The results for NSCLC and SCLC were reported separately. For patients with NSCLC, the study met its primary endpoint of improved immune-related progression-free survival (irPFS; as described in the guidelines for evaluation of immunotherapy activity [76]) for phased ipilimumab versus control (Fig. 3; hazard ratio (HR) = 0.72; p = 0.05), but not for concurrent ipilimumab (HR = 0.81; p = 0.13). Phased ipilimumab, concurrent ipilimumab, and control groups resulted in, respectively, median irPFS of 5.7, 5.5, and 4.6 months; an overall response rate (ORR) of 32, 21, and 14 %; and median OS of 12.2, 9.7, and 8.3 months. Likewise, compared with the control group, phased ipilimumab also significantly improved PFS according to modified WHO criteria (mWHO-PFS) (HR = 0.69; p = 0.02); however, the converse was true for concurrent ipilimumab (HR = 0.88; p = 0.25). Median mWHO-PFS for patients were treated with phased ipilimumab, concurrent ipilimumab, or control was 5.1, 4.1, and 4.2 months, respectively. When broken down according to histology, improvements in irPFS compared with controls seemed to be greater for patients with squamous cell carcinoma (HR = 0.55; 95 % confidence interval (CI), 0.27–1.12) than for patients with non-squamous cell tumors (HR = 0.82; 95 % CI, 0.52–1.28). Similar results were observed for WHO-modified PFS (squamous: HR = 0.40; 95 % CI, 0.18–0.87; non-squamous: HR = 0.81; 95 % CI, 0.53–1.26), and OS (squamous: 0.48; 95 % CI, 0.22–1.03; non-squamous: 1.17; 95 % CI, 0.74–1.86). However, there were no such differences based on histology for the concurrent ipilimumab group [74•].

Immune-related progression-free survival for phased (a), (c) and concurrent (b), (d) ipilimumab versus chemotherapy alone in patients with NSCLC and ED-SCLC [74•, 75] Reprinted with permission. © (2012) American Society of Clinical Oncology. All rights reserved. Lynch TJ, Bondarenko I, Luft A, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol. 2012;30:2046–2054; and reproduced by permission of Oxford University Press, Reck M, Bondarenko I, Luft A, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: results from a randomized, double-blind, multicenter phase 2 trial. Ann Oncol. 2012 Aug 2 (Epub ahead of print)
For the ED-SCLC cohort, phased ipilimumab, but not concurrent ipilimumab, improved irPFS versus control (HR = 0.64; p = 0.03), but no improvement in PFS or OS were observed [75]. Phased ipilimumab, concurrent ipilimumab, and control, respectively, were associated with median irPFS of 6.4, 5.7, and 5.3 months; median PFS of 5.2, 3.9, and 5.2 months; and median OS of 12.9, 9.1, and 9.9 months.
The incidence of the most common non-hematologic adverse events (AEs) typically associated with carboplatin and paclitaxel was similar across treatment groups and included fatigue, alopecia, nausea, vomiting, and peripheral sensory neuropathy. For other common AEs, including rash, pruritus, and diarrhea, there was a trend toward increased incidence in the ipilimumab groups. However, most AEs were grade 1 or 2. In the NSCLC cohort, the overall incidence of grade 3 and 4 immune-related AEs was 15, 20, and 6 % for the phased ipilimumab, concurrent ipilimumab, and control groups, respectively. Two patients (concurrent, n = 1; control, n = 1) died from treatment-related toxicity [74•]. In the ED-SCLC cohort, overall rates of grade 3 and 4 immune-related AEs were 17, 21, and 9 for phased ipilimumab, concurrent ipilimumab, and control, respectively [75].
The histology-specific results of this phase II study support further investigation of ipilimumab in two ongoing phase III trials. The first phase III trial is investigating the efficacy of ipilimumab or placebo in combination with carboplatin and paclitaxel in patients with stage IV or recurrent NSCLC of squamous histology (trial number NCT01285609). The second—a placebo-controlled trial—will assess the efficacy of ipilimumab in combination with platinum and/or etoposide chemotherapy for patients with newly diagnosed ED-SCLC (NCT01450761). An additional phase II study designed to investigate whether ipilimumab prolongs OS compared with pemetrexed for patients with recurrent or stage IV non-squamous NSCLC that has not progressed after four cycles of platinum-based first-line chemotherapy (NCT01471197) was closed early by the sponsor in mid-October 2012 because of the changing landscape associated with maintenance therapy.
A second checkpoint blockade strategy with antitumor activity is antibody-mediated targeting of the PD-1 receptor (by BMS-936558) or its ligand PD-L1 (BMS-936559, as discussed below). BMS-936558 (formerly MDX-1106) is a fully human anti-PD-1 mAb which binds to the PD-1 (or B7-H1) ligand, the predominant mediator of PD-1-dependent immune suppression. In murine models, B7-H1 expression confers immune resistance, and interrupting PD-1:B7-H1 expression has antitumor effects [61, 77, 78]. Results from a phase I dose-escalating study of BMS-936558 for patients with refractory solid tumors (compiled up to February 24, 2012) were recently reported [79]. In this publication, the cumulative ORR among 236 evaluable patients (assessed using modified response evaluation criteria in solid tumors (RECIST) version 1.0) was reported to be 18 % for NSCLC (14/76 patients), 28 % for melanoma (26/94 patients), and 27 % for renal cell carcinoma (9/33 patients) [79]. Notably, responses were durable and for patients with follow-up of more than one year, 20 of 31 responses lasted one year or more. Subsequent to the publication by Topalian et al. [79], data on the cohort of patients with NSCLC were updated to reflect longer follow-up, with approximately six additional months of patient accrual. These results, for a total of 127 patients with NSCLC, of whom more than 50 % had received ≥3 previous regimens, were presented at the 2012 European Society of Medical Oncology (ESMO) Annual Meeting [80••]. Responses were evaluable for 122 patients (48 squamous, 73 non-squamous, and 1 unknown histology). Tumor responses were observed for all three doses assessed (1, 3, and 10 mg kg−1 every two weeks) for squamous and non-squamous histology, with respective ORRs of 6, 27, and 17 %. Importantly, six patients with NSCLC had a non-conventional pattern of response and were not classified as responders by the conventional RECIST. Corresponding PFS at 24 weeks were 2, 44, and 31 %, respectively [80••]
Common treatment-related AEs observed in the total anti-PD-1-treated population included fatigue (24 %), reduced appetite (8 %), anemia (1 %), and nausea (8 %). The incidence of grade 3 and 4 treatment-related AEs was 14 % and there were three drug-related deaths from pneumonitis [79] (Table 1). A phase I study is in progress evaluating BMS-936558 in combination with three standard, platinum-based doublet chemotherapy regimens (cisplatin and gemcitabine, cisplatin and pemetrexed, and carboplatin and paclitaxel) for patients with treatment-naive stage IIIB or IV NSCLC (NCT01454102) [81]. The starting dose of BMS-936558 is 10 mg kg−1 every three weeks in each group, with a dose-expansion cohort in the carboplatin–paclitaxel group at 5 mg kg−1 every three weeks. Chemotherapy is given for four cycles whereas BMS-936558 is administered until disease progression, intolerable toxicity, or withdrawal of consent. The clinical potential of BMS-936558 is also being tested as second-line monotherapy. Two planned phase III studies are the first trials to compare “mono” immunotherapy with conventional second-line chemotherapy. These trials will compare BMS-936558 (3 mg kg−1 every two weeks) with docetaxel for patients with previously treated stage IIIB or IV NSCLC of, respectively, squamous-cell histology (NCT01642004) or non-squamous histology (NCT01673867). In both studies, the primary endpoint is OS, with ORR as an additional primary endpoint in the squamous cell study. The squamous-only trial has a co-primary endpoint of OS with ORR, whereas the primary endpoint in the non-squamous trial is OS only. A tumor sample is required as part of the inclusion criteria for both trials and clinical benefit—OS and ORR after treatment with BMS-936558 or docetaxel in PD-L1-positive versus PD-L1-negative expression subgroups—is a secondary endpoint in each trial.
As introduced above, BMS-936559 (a fully human IgGmAmAb) is another antigen-independent immunotherapy designed to regulate the PD-1 signaling system by targeting the PD-L1 ligand. Results from the first dose-escalating phase I trial for patients with relapsed or refractory solid tumors have been published recently (NCT00729664) [82, 83]. A total of 207 patients were enrolled, including melanoma (n = 55), NSCLC (n = 75), renal cell carcinoma (n = 17), and ovarian cancer (n = 17) patients. Clinical activity was observed for each of these four tumor types. Among the 49 evaluable patients with NSCLC, five patients achieved an objective response, of whom four were treated at the highest dose (10 mg kg−1) and responses were observed irrespective of histology. For all patients, the most common treatment-related AEs were fatigue, diarrhea, infusion reaction, arthralgia, rash, and pruritus.
In addition to checkpoint blockade, another antigen-independent immune modulation strategy is that of enhancing the immune priming process and abrogating immune tolerance of tumors by encouraging dendritic cell maturation in the presence of tumor antigens. To this end, talactoferrin has been assessed in clinical trials after evidence of antitumor activity against squamous cell and adenocarcinoma of the lung in animal models [71, 84, 85]. In a randomized phase II study, talactoferrin had activity against previously untreated stage IIIB or IV NSCLC in combination with paclitaxel and carboplatin, improving the response to chemotherapy alone (paclitaxel, carboplatin, and placebo) by 18 % for evaluable patients (29 to 47 %; p = 0.05) and by 15 % in the intent-to-treat population (27 to 42 %; p = 0.08) [86]. A separate randomized phase II study revealed that talactoferrin monotherapy statistically improved OS in the intent-to-treat population by 65 % (vs placebo) in NSCLC that had progressed after at least two previous systemic regimens, which supported phase III exploration of the agent in this setting [71]. In both of the randomized phase II trials, the incidence of AEs was lower in the talactoferrin arms than in the control arms [71, 86]. Despite a promising phase II experience, a recently completed randomized phase III study in advanced pretreated NSCLC reported no improvements in OS or PFS for talactoferrin over placebo, each in combination with best supportive care [87]. Investigation of talactoferrin continues for NSCLC (NCT00706862, in combination with carboplatin and paclitaxel) and for other tumor types (NCT00095186 and NCT01528137).
Clinical experience with antigen-specific vaccination therapy in NSCLC
Many efforts to investigate the clinical potential of targeting TAA in lung cancer have been completed or are in progress (Table 2). The MAGE-A3 vaccine, one therapeutic candidate, is composed of a recombinant fusion protein (MAGE-A3 and protein D of Haemophilus influenza) in combination with an immune-enhancing adjuvant [37]. The vaccine is currently being evaluated in the adjuvant setting after surgical resection of early-stage NSCLC. In a phase II trial, 182 patients with completely resected, MAGE-A3-positive, stage IB or II NSCLC were randomized 2:1 to MAGE-A3 vaccine or placebo [88, 89]. At the time of reporting, disease had recurred in 30.6 and 43.3 % of patients treated with vaccine and placebo, respectively. However, none of the outcome endpoints (disease-free interval, disease-free survival, or OS) reached statistical significance. The disease-free interval with the MAGE-A3 vaccine increased substantially (HR = 0.57) among patients with a gene expression signature possibly associated with a high risk of relapse [90]. The concept of antigen-specific cancer immunotherapy (ASCI) is currently being tested in the phase III double-blind trial MAGRIT (NCT00480025). In this study, patients with completely resected, pathologically proved stage IB, II, or IIIA MAGE-A3-positive NSCLC are randomly assigned 2:1 to receive a course of 13 injections of MAGE-A3 ASCI or placebo over a 27-month period, either directly after surgery or after surgery plus standard adjuvant chemotherapy. Recruitment for this trial was completed in December 2011. Over 1,300 patients were screened for MAGE-A3 expression, and 2,270 were randomized [37, 90]. Follow-up for the primary endpoint—disease-free survival—is in progress.
Another candidate under clinical investigation is the BLP25 liposomal vaccine (L-BLP25), a peptide antigen-based vaccine that targets the exposed core peptide of the MUC1-associated antigen. The vaccine includes the BLP25 lipopeptide and a liposomal delivery system, which facilitates uptake by APCs, and the immunoadjuvant monophosphoryl lipid A [91]. In preclinical studies of mice, the immune response to L-BLP25 was characterized by a proliferative T cell response to the MUC1 antigen and production of IFN-γ [92]. A phase I/II trial for patients with NSCLC found no significant safety issues and showed that L-BLP25 was capable of eliciting a T cell response [93]. Another investigation compared the efficacy of L-BLP25 plus best supportive care (BSC) with BSC alone in a phase II trial of 171 patients with stage IIIB or IV NSCLC that was stable or had responded to any first-line chemotherapy [91, 94]. Patients in the L-BLP25 group received a single low dose of cyclophosphamide (shown to enhance the effect of immunotherapy) three days before the first dose of L-BLP25, followed by eight weekly subcutaneous vaccinations of 1-mg L-BLP25, and maintenance vaccinations every six weeks thereafter. In updated survival analysis, median OS was 4.2 months longer in the vaccine plus BSC group (17.2 months) than in the BSC alone group (13 months)—a trend that did not reach significance (HR = 0.745; 95 % CI 0.533–1.042) [94]. Three-year survival was 31 % in the L-BLP25 plus BSC group compared with 17 % in the BSC only group (p = 0.035). In the stratified subset of patients with stage IIIB loco-regional disease, OS was improved for patients receiving L-BLP25 plus BSC compared with those receiving BSC only (median 30.6 months vs. 13.3 months, respectively; HR = 0.548; 95 % CI 0.301–0.999). In this subgroup, three-year survival was 49 and 27 % in the L-BLP25 plus BSC and BSC only groups, respectively (p = 0.070) [94]. On the basis of these results, two phase III L-BLP25 trials have been started for patients with unresectable stage III NSCLC. The trial design is the same as that for the phase II trial, with the notable exceptions being that both trials are double-blind, placebo-controlled and that the phase III trials include only patients with unresectable stage III NSCLC whereas the phase II trial included patients with stage IIIB (locoregional or stage IIIB with malignant pleural effusion) and stage IV disease. In each case, OS is the primary endpoint. The START trial (NCT00409188) has completed recruitment and reached its accrual target of nearly 1,500 patients, whereas the INSPIRE trial (NCT01015443) will accrue 420 patients from East Asia.
The TG4010 vaccine also targets the MUC1 antigen. TG4010 consists of a suspension of attenuated Ankara virus which has been genetically modified to express MUC1 and IL-2 [95]; IL-2 acts as a strong immunoadjuvant, capable of reversing the suppression of the T-cell response mediated by cancer-associated MUC1 [96]. A randomized phase IIB study suggested that TG4010 enhanced the effect of chemotherapy (cisplatin plus gemcitabine) for 148 patients with stage IIIB or IV MUC1-positive NSCLC [97]. PFS at six months was 43.2 % (95 % CI 33.4–53.5) in the TG4010 plus chemotherapy group and 35.1 % (95 % CI 25.9–45.3) in the chemotherapy alone group. AEs that were more common in the TG4010 group than the chemotherapy-alone group included fever (23 % versus 8 %), abdominal pain (16 % versus 3 %) and injection-site pain (5 % versus 0 %). A confirmatory, double-blind, placebo-controlled phase IIB/III trial is in progress to investigate the efficacy and safety of first-line TG4010 combined with chemotherapy for patients with MUC1-positive stage IV NSCLC who have normal baseline NK levels (NCT01383148). The primary endpoints are PFS and OS for the phase II and III stages, respectively.
The belagenpumatucel-L vaccine (Lucanix™) is an allogeneic cell vaccine comprising four different NSCLC cell lines (two adenocarcinoma, one squamous cell carcinoma, and one large cell carcinoma) and therefore includes a large number of antigens. The NSCLC cell lines are transfected with a TGF-β2 antisense gene which downregulates TGF-β2 and increases the immunogenicity of the transformed cells [98]. In a randomized, dose-variable phase II trial for 75 patients with stages II, IIIA, IIIB, and IV NSCLC, patients received one of three doses of belagenpumatucel-L on a monthly or every-other-month schedule, for a maximum of 16 injections [98]. Among the 61 patients with stage IIIB or IV disease, the ORR was 15 %. One and two-year survival in the higher dose groups combined was 68 and 52 %, respectively; it was 39 and 20 % respectively in the low-dose group. The STOP phase III trial compares belagenpumatucel-L with placebo as maintenance therapy for patients with stage III or IV NSCLC that is stable after, or has responded to, first-line platinum-based chemotherapy (NCT00676507). In this study, patients receive belagenpumatucel-L (or placebo) intradermally once monthly for 18 months, and then once after 21 and 24 months in the absence of disease progression or unacceptable toxicity. The primary endpoint is OS, and the trial recently achieved its planned recruitment of approximately 506 patients.
While in existence for some time, an EGF vaccine previously developed in Cuba comprising recombinant human EGF coupled to a carrier protein, with aluminium hydroxide or containing ISA51 as immunoadjuvant, has been the subject of recent scrutiny in lung cancer [99]. In a phase II study, 80 patients with stage IIIB or IV NSCLC were randomized to receive EGF vaccination or BSC [100]. As with L-BLP25, vaccination was primed with cyclophosphamide and the EGF vaccine given on days 1, 7, 14, and 28, and monthly thereafter. Median OS in the EGF vaccine and BSC groups was 6.5 and 5.3 months, respectively (not significant); in subgroup analysis of patients younger than 60 years, median OS was 11.6 and 5.3 months (p = 0.0124). Immune response seemed to predict treatment benefit; those with a good antibody response had a median OS of 11.7 months compared with 3.6 months for others (p = 0.002). An international phase III trial comparing EGF vaccination with BSC for patients with stage IIIB and IV NSCLC is in progress (NCT01444118), with OS as the primary endpoint.
Unanswered questions and future directions
Despite clinical application of new chemotherapeutic agents and molecularly targeted therapy, patient outcomes have remained unfavorable for NSCLC—a histologic subtype of lung cancer which accounts for nearly 85 % of all cases. As such, the development of novel, efficacious, and tolerable therapy with clinical applicability is warranted. Today, enhanced understanding of the mechanisms of immune-mediated elimination of tumor cells has created a niche for immunotherapy as a treatment option for lung cancer. In this review we discussed clinical experience with two approaches to immunotherapy for lung cancer—antigen-independent immunomodulatory therapy and antigen-specific cancer immunotherapy. For both approaches, favorable of efficacy and safety outcomes have been observed in phase II trials, and they are currently being investigated in phase III studies. However, a better understanding of the specific population of patients that will benefit from such therapy is imperative, as is a better conceptual grasp of other aspects of therapeutic strategy.
Some questions that remain unanswered and are thus ideal topics for future study include recognizing the outcomes of therapy co-administered with immunotherapy. For example, what are the interactions between immunotherapy and chemotherapy, or immunotherapy and radiotherapy? Similarly, are there any potential interactions between antigen-independent and antigen-specific therapy, and do results support the rationale to combine the two? And, importantly, how should such therapy be co-administered with regard to timing and sequencing? When pulmonologists and oncologists measure the efficacy of immunotherapy, what is the best method available? Are the immune-response criteria devised especially for immunotherapy—to differentiate responses observed after treatment with immunotherapy from those that result from chemotherapy—sufficient?
As noted above, selecting the most appropriate patient population remains a challenge and, thus, determining the best biomarkers to use to facilitate determination of prognosis, efficacy, and immunogenicity would be very beneficial. When patients have been treated with the appropriate immunotherapy, how should clinicians define the role of maintenance therapy? Is it truly efficacious? And in the context of oncogene inhibitors, how do you explain the different paradigm observed? Another unknown faced by clinicians who administer immunotherapy is whether the efficacy of the new treatment approach could be assessed for patients with advanced tumor disease, or would it be something that could only be assessed for patients with relatively minor tumor burden? What strategies should clinicians use to reach a conclusion? Further studies of immunotherapy in some of the different patient populations described are both necessary and germane to optimization of newer treatment strategies for patients with lung cancer.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Ferlay J, Shin H-R, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–917.
Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367–80.
American Cancer Society. Available from: http://www.cancer.org/Cancer/LungCancer-Non-SmallCell/OverviewGuide/lung-cancer-non-small-cell-overview-key-statistics. Accessed August 31, 2012.
NCCN Guidelines® Version 3.2012. Non-small cell lung cancer. Available at http://www.nccn.org/professionals/physician_gls/f_guidelines.asp#site. Accessed August 31, 2012.
Brodowicz T, Ciuleanu T, Crawford J, et al. Third CECOG consensus on the systemic treatment of non-small-cell lung cancer. Ann Oncol. 2012;23:1223–9.
D’Addario G, Felip E, ESMO Guidelines Working Group. Non-small-cell lung cancer. ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2009;20 suppl 4:68–70.
Shepherd F, Douillard J-Y, Blumenschein GR. Immunotherapy for non-small-cell lung cancer: novel approaches to improve patient outcome. J Thorac Oncol. 2011;6:1763–73.
Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–71.
Engel AM, Svane IM, Mouritsen S, Rygaard J, Clausen J, Werdelin O. Methylcholanthrene induced sarcomas in nude mice have short induction times and relatively low levels of surface MHC class I expression. APMIS. 1996;104:629–39.
Engel AM, Svane IM, Rygaard J, Werdelin O. MCA sarcomas induced in scid mice are more immunogenic than MCA sarcomas induced in congenic, immunocompetent mice. Scand J Immunol. 1997;45:463–70.
Shankaran V, Ikeda H, Bruce AT, et al. IFNγ and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–11.
Smyth MJ, Crowe NY, Godfrey DI. NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. Int Immunol. 2001;13:459–6.
Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011;331:1565–70.
Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30:1073–81.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–8.
Koebel CM, Vermi W, Swann JB, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007;450:903–7.
Eyles J, Puaux AL, Wang X, et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J Clin Invest. 2010;120:2030–9.
Hurwitz AA, Watkins SK. Immune suppression in the tumor microenvironment: a role for dendritic cell-mediated tolerization of T cells. Cancer Immunol Immunother. 2012;61:289–93.
Ganss R, Hanahan D. Tumor microenvironment can restrict the effectiveness of activated antitumor lymphocytes. Cancer Res. 1998;58:4673–81.
Anderson MJ, Shafer-Weaver K, Greenberg NM, Hurwitz AA. Tolerization of tumor-specific T cells despite efficient initial priming in a primary murine model of prostate cancer. J Immunol. 2007;178:1268–76.
The Board of Trustees of the Leland Stanford Junior University. Immune Evasion. Parasites and Pestilence. 2007. Available from: http://www.stanford.edu/class/humbio153/ImmuneEvasion/Background.html.
Hiraoka K, Miyamoto M, Cho Y, et al. Concurrent infiltration by CD8+ T cells and CD4+ T cells is a favourable prognostic factor in non-small-cell lung carcinoma. Br J Cancer. 2006;94:275–80.
Al-Shibli KI, Donnem T, Al-Saad S, Persson M, Bremnes RM, Busund LT. Prognostic effect of epithelial and stromal lymphocyte infiltration in non-small-cell lung cancer. Clin Cancer Res. 2008;14:5220–7.
Zhuang X, Xia X, Wang C, et al. A high number of CD8+ T cells infiltrated in NSCLC tissues is associated with a favorable prognosis. Appl Immunohistochem Mol Morphol. 2010;18:24–8.
Kawai O, Ishii G, Kubota K, et al. Predominant infiltration of macrophages and CD8(+) T cells in cancer nests is a significant predictor of survival in stage IV nonsmall-cell lung cancer. Cancer. 2008;113:1387–95.
Dai F, Liu L, Che G, et al. The number and microlocalization of tumor-associated immune cells are associated with patient's survival time in non-small-cell lung cancer. BMC Cancer. 2010;10:220.
Okita R, Saeki T, Takashima S, Yamaguchi Y, Toge T. CD4+CD25+ regulatory T cells in the peripheral blood of patients with breast cancer and non-small-cell lung cancer. Oncol Rep. 2005;14:1269–73.
Karagöz B, Bilgi O, Gümüs M, et al. CD8+CD28- cells and CD4+CD25+ regulatory T cells in the peripheral blood of advanced stage lung cancer patients. Med Oncol. 2010;27:29–33.
Wang YY, He XY, Cai YY, Wang ZJ, Lu SH. The variation of CD4+CD25+ regulatory T cells in the periphery blood and tumor microenvironment of non-small-cell lung cancer patients and the downregulation effects induced by CpG ODN. Target Oncol. 2011;6:147–54.
Erfani N, Mehrabadi SM, Ghayumi MA, et al. Increase of regulatory T cells in metastatic stage and CTLA-4 over expression in lymphocytes of patients with non-small-cell lung cancer (NSCLC). Lung Cancer. 2012;77:306–11.
Ju S, Qiu H, Zhou X, et al. CD13+CD4+CD25hi regulatory T cells exhibit higher suppressive function and increase with tumor stage in non-small-cell lung cancer patients. Cell Cycle. 2009;8:2578–85.
Petersen RP, Campa MJ, Sperlazza J, et al. Tumor infiltrating Foxp3+ regulatory T-cells are associated with recurrence in pathologic stage I NSCLC patients. Cancer. 2006;107:2866–72.
Shimizu K, Nakata M, Hirami Y, Yukawa T, Maeda A, Tanemoto K. Tumor-infiltrating Foxp3+ regulatory T cells are correlated with cyclooxygenase-2 expression and are associated with recurrence in resected non-small-cell lung cancer. J Thorac Oncol. 2010;5:585–90.
Tao H, Mimura Y, Aoe K, et al. Prognostic potential of FOXP3 expression in non-small-cell lung cancer cells combined with tumor-infiltrating regulatory T cells. Lung Cancer. 2012;75:95–101.
Liu H, Zhang T, Ye J, et al. Tumor-infiltrating lymphocytes predict response to chemotherapy in patients with advance non-small-cell lung cancer. Cancer Immunol Immunother. 2012 Mar 29. [Epub ahead of print]
Decoster L, Wauters I, Vansteenkiste JF. Vaccination therapy for non-small-cell lung cancer: review of agents in phase III development. Ann Oncol. 2012;23:1387–93.
Situ D, Wang J, Ma Y, et al. Expression and prognostic relevance of MUC1 in stage IB non-small-cell lung cancer. Med Oncol. 2011;28 suppl 1:S596–604.
Sienel W, Varwerk C, Linder A, et al. Melanoma associated antigen (MAGE)-A3 expression in stages I and II non-small-cell lung cancer: results of a multi-center study. Eur J Cardiothorac Surg. 2004;25:131–4.
Kim SH, Lee S, Lee CH, et al. Expression of cancer-testis antigens MAGE-A3/6 and NY-ESO-1 in non-small-cell lung carcinomas and their relationship with immune cell infiltration. Lung. 2009;187(6):401–11.
Shigematsu Y, Hanagiri T, Shiota H, et al. Clinical significance of cancer/testis antigens expression in patients with non-small-cell lung cancer. Lung Cancer. 2010;68:105–10.
Grah J, Samija M, Juretić A, Sarcević B, Sobat H. Immunohystochemical expression of cancer/testis antigens (MAGE-A3/4, NY-ESO-1) in non-small-cell lung cancer: the relationship with clinical-pathological features. Coll Antropol. 2008;32:731–6.
Gure AO, Chua R, Williamson B, et al. Cancer-testis genes are coordinately expressed and are markers of poor outcome in non-small-cell lung cancer. Clin Cancer Res. 2005;11:8055–62.
Fernandez LE, Gabri MR, Guthmann MD, et al. NGcGM3 ganglioside: a privileged target for cancer vaccines. Clin Dev Immunol. 2010;2010:814397.
Linsley PS, Greene JL, Tan P, et al. Coexpression and functional cooperation of CTLA-4 and CD28 on activated T lymphocytes. J Exp Med. 1992;176(6):1595–604.
Walunas TL, Lenschow DJ, Bakker CY, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–13.
Hoos A, Ibrahim R, Korman A, et al. Development of ipilimumab: contribution to a new paradigm for cancer immunotherapy. Semin Oncol. 2010;37:533–46.
Salama AKS, Hodi FS. Cytotoxic T-lymphocyte-associated antigen-4. Clin Cancer Res. 2011;17:4622–8.
Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459–65.
Krummel MF, Sullivan TJ, Allison JP. Superantigen responses and co-stimulation: CD28 and CTLA-4 have opposing effects on T cell expansion in vitro and in vivo. Int Immunol. 1996;8(4):519–23.
Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3(5):541–7.
Waterhouse P, Penninger JM, Timms E, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–8.
Fife BT, Pauken KE. The role of the PD-1 pathway in autoimmunity and peripheral tolerance. Ann N Y Acad Sci. 2011;1217:45–59.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;22;12(4):252–64.
Sapoznik S, Hammer O, Ortenberg R, et al. Novel anti-melanoma immunotherapies: disarming tumor escape mechanisms. Clin Dev Immunol. 2012;2012:818214.
Nazareth MR, Broderick L, Simpson-Abelson MR, Kelleher RJ, Yokota SJ, Bankert RB. Characterization of human lung tumor-associated fibroblasts and their ability to modulate the activation of tumor-associated T cells. J Immunol. 2007;178:5552–62.
Pinchuk IV, Saada JI, Beswick EJ, et al. PD-1 ligand expression by human colonic myofibroblasts/fibroblasts regulates CD4+ T-cell activity. Gastroenterology. 2008;135:1228–37.
Ohigashi Y, Sho M, Yamada Y, et al. Clinical significance of programmed death-1 ligand-1 and programmed death-1 ligand-2 expression in human esophageal cancer. Clin Cancer Res. 2005;11:2947–53.
Liang SC, Latchman YE, Buhlmann JE, et al. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur J Immunol. 2003;33:2706–16.
Okazaki T, Honjo T. The PD-1-PD-L pathway in immunological tolerance. Trend Immunol. 2006;27:195–201.
Iwai Y, Ishida M, Tanaka Y, et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002;99:12293–7.
Zhang L, Gajewski TF, Kline J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood. 2009;114:1545–52.
Iwai Y, Terawaki S, Honjo J. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int Immunol. 2005;17:133–44.
Iwai Y, Terawaki S, Ikegawa M, Okazaki T, Honjo T. PD-1 inhibits antiviral immunity at the effector phase in the liver. J Exp Med. 2003;198:39–50.
Perrot I, Blanchard D, Freymond N, et al. Dendritic cells infiltrating human non-small-cell lung cancer are blocked at immature stage. J Immunol. 2007;178(5):2763–9.
Tabarkiewicz J, Rybojad P, Jablonka A, Rolinski J. CD1c+ and CD303+ dendritic cells in peripheral blood, lymph nodes and tumor tissue of patients with non-small-cell lung cancer. Oncol Rep. 2008;19:237–43.
Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–48.
Schneider T, Hoffmann H, Dienemann H, et al. Non-small-cell lung cancer induces an immunosuppressive phenotype of dendritic cells in tumor microenvironment by upregulating B7-H3. J Thorac Oncol. 2011;6:1162–8.
Loos M, Hedderich DM, Friess H, Kleef J. B7-h3 and its role in antitumor immunity. Clin Dev Immunol. 2010;2010:683875.
Spadaro M, Caorsi C, Ceruti P, et al. Lactoferrin, a major defense protein of innate immunity, is a novel maturation factor for human dendritic cells. FASEB J. 2008;22(8):2747–57.
Parikh PM, Vaid A, Advani SH, et al. Randomized, double-blind, placebo-controlled phase II study of single-agent oral talactoferrin in patients with locally advanced or metastatic non–small-cell lung cancer that progressed after chemotherapy. J Clin Oncol. 2011;29:4129–36.
Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23.
Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–26.
• Lynch TJ, Bondarenko I, Luft A, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol. 2012;30:2046–54. This trial was the first to demonstrate a clinical benefit for adding CTLA-4 blockade, via ipilimumab, to chemotherapy as a treatment for patients with advanced NSCLC.
Reck M, Bondarenko I, Luft A, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: results from a randomized, double-blind, multicenter phase 2 trial. Ann Oncol. 2012 Aug 2. [Epub ahead of print].
Wolchok JD, Hoos A, O'Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15:7412–20.
Strome SE, Dong H, Tamura H, et al. B7-H1 blockade augments T-cell immunotherapy for squamous cell carcinoma. Cancer Res. 2003;63:6501–5.
Blank C, Brown I, Peterson AC, et al. PD-L1/ B7-H1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8_ T cells. Cancer Res. 2001;64:1140–5.
Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1antibody in cancer. N Engl J Med. 2012;366:2443–54.
•• Gettinger S, Horn L, Antonia SJ, et al. Clinical activity and safety of anti-programmed death-1 (PD-1) (BMS-936558/MDX-1106/ONO-4538) in patients with advanced non-small-cell lung cancer (NSCLC). Ann Oncol. 2012a;23(suppl 9):abstr 1237PD. This trial identifies clinical activity of PD-1 blockade, via BMS-936558, against advanced NSCLC.
Gettinger SN, Rizvi NA, Shepherd FA, et al. A phase I study of BMS-936558 in combination with gemcitabine/cisplatin, pemetrexed/cisplatin, or carboplatin/paclitaxel in patients with treatment-naïve, stage IIIB/IV non-small-cell lung cancer. J Clin Oncol. 2012b;30(suppl):abstr TPS2615.
Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65.
Tykodi SS, Brahmer JR, Hwu W-J, et al. PD-1/PD-L1 pathway as a target for cancer immunotherapy: safety and clinical activity of BMS-936559, an anti-PD-L1 antibody, in patients with solid tumors. J Clin Oncol. 2012;30:(suppl; abstr 2510).
Spadaro M, Curcio C, Varadhachary A, et al. Requirement for IFN-gamma, CD8+ T lymphocytes, and NK T cells in talactoferrin-induced inhibition of neu+ tumors. Cancer Res. 2007;67:6425–32.
Varadhachary Varadhachary A, Wolf JS, Petrak K, et al. Oral lactoferrin inhibits the growth of established tumors and potentiates conventional chemotherapy. Int J Cancer. 2004;111:398–403.
Digumarti R, Wang Y, Raman G, et al. A randomized, double-blind, placebo-controlled, phase II study of oral talactoferrin in combination with carboplatin and paclitaxel in previously untreated locally advanced or metastatic non-small-cell lung cancer. J Thorac Oncol. 2011;6(6):1098–103.
Ramalingam SS, Crawford J, Chang A, et al. FORTIS-M, a randomized, double-blind, placebo-controlled phase 3 study of oral talactoferrin alfa with best supportive care in patients with advanced non-small-cell lung cancer following two or more prior regimens- by The FORTIS-M Study Group. Presented at the 2012 ESMO meeting; abstr. LBA34.
Vansteenkiste JF, Zielinski M, Linder A, et al. Final results of a multi-center, double-blind, randomized, placebo-controlled phase II study to assess the efficacy of MAGE-A3 immunotherapeutic as adjuvant therapy in stage IB/II non-small-cell lung cancer (NSCLC). J Clin Oncol. 2007;25:(June 20 suppl; abstr 7554).
Bradbury PA, Shepherd FA. Immunotherapy for lung cancer. J Thoracic Cancer. 2008;3 suppl 2:S164–70.
Vansteenkiste JF, Zielinski M, Dahabreh IJ, et al. Association of gene expression signature and clinical efficacy of MAGE-A3 antigen-specific cancer immunotherapeutic (ASCI) as adjuvant therapy in resected stage IB/II non-small-cell lung cancer (NSCLC). J Clin Oncol 2008;26:(May 20 suppl; abstr 7501).
Butts C, Murray N, Maksymiuk A, et al. Randomized phase IIB trial of BLP25 liposome vaccine in stage IIIB and IV non-small-cell lung cancer. J Clin Oncol. 2005;23:6674–81.
Guan HH, Budzynski W, Koganty RR, et al. Liposomal formulations of synthetic MUC1 peptides: effects of encapsulation versus surface display of peptides on immune responses. Bioconjug Chem. 1998;9:451–8.
Palmer M, Parker J, Modi S, et al. Phase I study of the BLP25 (MUC1 peptide) liposomal vaccine for active specific immunotherapy in stage IIIB/IV non-small-cell lung cancer. Clin Lung Cancer. 2001;3:49–57.
Butts C, Maksymiuk A, Goss G, et al. Updated survival analysis in patients with stage IIIB or IV non-small-cell lung cancer receiving BLP25 liposome vaccine (L-BLP25): phase IIB randomized, multicenter, open-label trial. J Cancer Res Clin Oncol. 2011;137:1337–42.
Ramlau R, Quoix E, Rolski J, et al. A phase II study of Tg4010 (Mva-Muc1-Il2) in association with chemotherapy in patients with stage III/IV non-small-cell lung cancer. J Thorac Oncol. 2008;3:735–44. Erratum in: J Thorac Oncol. 2008;3:941.
Agrawal B, Krantz MJ, Reddish MA, Longenecker BM. Cancer-associated MUC1 mucin inhibits human T-cell proliferation, which is reversible by IL-2. Nat Med. 1998;4:43–9.
Quoix E, Ramlau R, Westeel V, et al. Therapeutic vaccination with TG4010 and first-line chemotherapy in advanced non-small-cell lung cancer: a controlled phase 2B trial. Lancet Oncol. 2011;12:1125–33.
Nemunaitis J, Dillman RO, Schwarzenberger PO, et al. Phase II study of belagenpumatucel-L, a transforming growth factor beta-2 antisense gene-modified allogeneic tumor cell vaccine in non-small-cell lung cancer. J Clin Oncol. 2006;24:4721–30.
González G, Crombet T, Catalá M, Mirabal V, Hernández JC, González Y, et al. A novel cancer vaccine composed of human-recombinant epidermal growth factor linked to a carrier protein: report of a pilot clinical trial. Ann Oncol. 1998;9:431–5.
Neninger Vinageras E, de la Torre A, Osorio Rodríguez M, et al. Phase II randomized controlled trial of an epidermal growth factor vaccine in advanced non-small-cell lung cancer. J Clin Oncol. 2008;26:1452–8.
Acknowledgments
Professional medical writing and editorial assistance was provided by StemScientific. Funding for this assistance was provided by Bristol–Myers–Squibb.
Disclosure
M. Reck: consultant to Hoffmann–La Roche, Lilly, Daiichi–Sankyo, BMS, and AstraZeneca and speakers’ bureaus for Hoffmann–La Roche, Lilly, Daiichi–Sankyo, AstraZeneca, and Pfizer; J. Vansteenkiste: none; and J. R. Brahmer: consultant to Bristol–Myers–Squibb, Merck, Eli Lilly, and Cephalon.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Reck, M., Vansteenkiste, J. & Brahmer, J.R. Targeting the immune system for management of NSCLC: the revival?. Curr Respir Care Rep 2, 22–39 (2013). https://doi.org/10.1007/s13665-012-0038-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13665-012-0038-5