
Abstract
Two hitherto unknown lanostane-type triterpenoids, namely scillascillol (1) and scillascillone (2), and a hitherto unknown norlanostane-triterpene glycoside, namely scillascilloside B-1 (3), were isolated from the ethanol extract of the whole plants of Scilla scilloides. Their structures were elucidated on the basis of extensive spectroscopic studies. In addition, the structure of drimiopsin D (6a) has been revised as 2,5-dimethoxy-8-methyl-1,3,6-trihydroxyxanthone (6) by reanalysis of the spectroscopic data.
Graphical Abstract

Similar content being viewed by others


1 Introduction
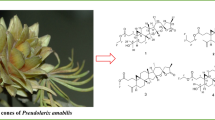
Scilla scilloides (Lindl.) Druce is a perennial herb belonging to the Liliaceae family, also compiled in Hyacinthaceae family in the relatively new classification system [1]. The bulbs or the whole plants have been used as a foodstuff, a traditional Chinese medicine for promoting blood circulation, an analgesic, an anti-inflammatory agent, and treatment of heart failure and arrhythmia [2]. Previous chemical investigations on S. scilloides reported homoisoflavones, lanostane-type and norlanostane-type triterpenoids, xanthones, lignans, etc. [3–10]. As part of a BioBioPha (http://www.chemlib.cn) objective to assemble a large-scale natural product library valuable in the discovery of new drug leads from nature [11–14], the phytochemical investigation on the whole plants of S. scilloides led to isolation of two new lanostane-type triterpenoids, namely scillascillol (1) and scillascillone (2), a new norlanostane-triterpene glycoside, namely scillascilloside B-1 (3), together with two known norlanostanes, 15-deoxoeucosterol (4) [4] and 3-dehydro-15-deoxoeucosterol (5) [4], and three known xanthones, drimiopsin D (6) [15], drimiopsin C (7) [15] and norlichexanthone (8) [16] (Fig. 1). This paper describes the isolation and structural elucidation of new lanostane-type triterpenoids and structure revision of a xanthone drimiopsin D.

Structures of compounds 1–8
2 Results and Discussion
Compound 1 was obtained as a white amorphous powder. Its molecular formula was determined to be C30H46O6 on the basis of negative-ion HRESIMS at m/z 501.3207 [M − H]− (calcd for C30H45O6, 501.3216). The IR absorption bands at 3441, 3432 and 1765 cm−1 suggested the presence of hydroxy and carbonyl functionalities, respectively. The 1H NMR spectrum (Table 1) of 1 indicated signals due to four tertiary methyls [δ H 0.89 (s), 1.01 (s), 1.24 (s), 1.52 (s)], two secondary methyls [δ H 1.07 (d, J = 6.8 Hz); 1.47 (d, J = 7.1 Hz)], a pair of oxygenated methylene protons [δ H 3.70, 4.56 (each d, J = 11.0 Hz)], and two oxygenated methine protons [δ H 3.61 (dd, J = 11.6, 4.4 Hz); 4.34 (d, J = 4.4 Hz)]. The 13C NMR and DEPT spectrum (Table 1) exhibited 30 carbon signals, including an ester carbonyl resonance at [δ C 178.9 (s)], a tetra-substituted double bond at [δ C 135.1 (s), 134.9 (s)], and five oxygenated carbons at [δ C 64.5 (t), 77.4 (d), 80.0 (d), 99.2 (s), 117.0 (s)]. By analyzing the above signals, especially the set of particular quaternary carbons [δ C 99.2, 117.0, 178.9] and an oxygenated methine [δ H 4.34 (d, J = 4.4 Hz); δ C 77.4 (d)], compound 1 was presumed as a kind of characteristic lanostane triterpenoid derived from this genus. The above NMR spectroscopic features of 1 were very similar to those of scillasaponin D [17] and scillasaponin G [18], except lack of the signals due to the sugar moiety and an up-field shift about 10 ppm of the 13C NMR signal at C-3 of 1. Based on analysis of the above spectral data, a planar structure of 1 was assumed, which was also confirmed by the HMBC (Fig. 2) correlations from H-3 to C-1 and C-29, from Me-19 to C-1 and C-9, as well as from Me-28 and H-29 to C-3. Taking into account its biological source and characteristic NMR data (including chemical shifts and coupling constants) of the spiro rings, the stereochemistry of 1 should be consistent with those analogues reported from this genus [8]. The hydroxy group at C-3 was equatorial (β) according to an axial–axial coupling between H-3 and H-2β (J = 11.6 Hz) and an axial-equatorial coupling between H-3 and H-2α (J = 4.4 Hz), which was also supported by the ROESY (Fig. 2) correlations of H-3 ↔ H-5 and H-3 ↔ Me-28. Considering a relatively good rigid property of spiro rings, the determination of its stereochemistry was able to be conducted on the basis of ROESY analysis. The ROESY correlations of H-24 ↔ Me-21, H-25 ↔ Me-30 and Me-30 ↔ H-15α were indicative of α-orientation of these protons, while the correlations of Me-18 ↔ H-20/H-11β indicative of their β-orientation. Therefore, the structure of 1 was determined as 17α,23α-epoxy-3β,24β,29-trihydroxylanost-8-en-26,23-olide and named scillascillol.

Key HMBC ( ) and ROESY (
) and ROESY ( ) correlations of 1
) correlations of 1
Compound 2 was isolated as a white amorphous powder. The HRESIMS (neg.) at m/z 499.3053 [M − H]− (calcd for C30H43O6, 499.3060) indicated the molecular formula C30H44O6, corresponding to nine degrees of unsaturation. By comparison with the reported NMR data [17, 18], the signals of characteristic five-membered spiro rings, a tetra-substituted double bond and six methyls were detected as before, which indicated that 2 had a very similar structure to scillascillol (1). The main difference is only that 2 had a newly arising keto carbon [δ C 214.5 (s)] instead of the oxygenated methine signal [δ H 3.61 (dd, J = 11.6, 4.4 Hz); δ C 80.0 (d)] at C-3 of 1. Thus, compound 2 should be the 3-dehydro derivative of 1. And this was confirmed by the HMBC correlations from H-1 to C-3, C-5, C-9 and C-10, as well as from Me-28 and H-29 to C-3, C-4 and C-5. Therefore, the structure of 2 was determined as 24β,29-dihydroxy-17α,23α-epoxy-3-oxolanost-8-en-26,23-olide and named scillascillone.
Compound 3 was also obtained as a white amorphous powder, and its molecular formula was deduced to be C40H64O13 based on its negative-ion HRESIMS at m/z 751.4261 [M − H]− (calcd for C40H63O13, 751.4269). The 1H NMR spectrum (Table 2) of 3 was characterized by signals due to four tertiary methyls [δ H 1.48 (s), 1.47 (s), 0.90 (s), 0.87 (s)], one secondary methyl [δ H 1.00 (d, J = 6.6 Hz)], one primary methyl [δ H 1.05 (t, J = 7.3 Hz)], and two anomeric protons [δ H 4.98 (d, J = 7.9 Hz); 4.92 (d, J = 7.0 Hz)]. The 13C NMR spectrum (Table 2) of 3 implied the presence of a tetra-substituted double bond [δ C 135.2 (s), 134.6 (s)], one keto carbonyl [δ C 212.6 (s)], and two anomeric carbons [δ C 106.2 (d), 105.6 (d)]. On the other hand, a set of additional 11 oxygenated carbons were detected by comparison with those of 15-deoxoeucosterol (4), suggesting the presence of a hexose and a pentose. The assignment of the hexose as C-6 glycosylated glucopyranosyl was launched by observation of a set of specific oxygenated carbons at [δ C 106.2 (d), 75.4 (d), 78.7 (d), 72.0 (d), 77.1 (d), 69.9 (t)] and a diagnostic glycosidation shift (∆ ≈ +8 ppm) of C-6 of glucopyranosyl. It was still supported by the detailed HMBC analysis and comparison with the reported spectral data [19]. The remaining carbons at [δ C 105.6 (d), 72.3 (d), 74.4 (d), 69.3 (d), 66.7 (t)] were established as a terminal α-l-arabinopyranosyl unit by analysis of coupling constants of sugar moiety (Fig. 3), in which axial–axial couplings between H-1 and H-2 (J = 7.0 Hz), and between H-2 and H-3 (J = 8.5 Hz), and axial-equatorial coupling between H-3 and H-4 (J = 2.8 Hz) were clearly observed. And the above-mentioned NMR feature was also consistent with the reported spectral data [20, 21]. In addition, on acid hydrolysis, 3 afforded monosaccharides in the aqueous layer, which were found to be identical with authentic samples of l-arabinose and d-glucose by chiral HPLC analysis. The connection of these units was established by the HMBC correlations from the anomeric proton of the arabinopyranosyl at [δ H 4.92 (d, J = 7.0 Hz)] to C-6 of the glucopyranosyl at [δ C 69.9 (t)], and from the anomeric proton of the glucopyranosyl at [δ H 4.98 (d, J = 7.9 Hz)] to C-3 of the aglycone at [δ C 88.9 (d)]. Thus, compound 3 was concluded to be 15-deoxoeucosterol 3-O-α-l-arabinopyranosyl-(1 → 6)-β-d-glucopyranoside, and named scillascilloside B-1.

Chair conformation of sugar moiety in 3
Compound 6, a yellow amorphous powder, had a molecular formula of C16H14O7 based on HRESIMS (neg.) at m/z 317.0665 [M − H]− (calcd for C16H13O7, 317.0661). The 1H NMR spectrum (Table 3) indicated the presence of two methoxy signals [δ H 3.72 (s), 3.81 (s)], one aromatic methyl [δ H 2.66 (s)], two aromatic singlets [δ H 6.41, 6.69], and three low-field exchangeable proton singlets at [δ H 10.73, 10.79, 13.44]. The 13C NMR spectrum (Table 3) displayed a total of 16 carbon resonances ascribable to two benzene rings, two methoxy group [δ C 60.0 (q), 60.8 (q)], one methyl [δ C 22.8 (q)], and one carbonyl carbon [δ C 182.1 (s)]. The above NMR spectroscopic features were very similar to those of drimiopsin I [22], and the most significant difference was from an additional methoxy signal in 6. The methoxy group was positioned at C-2 according to the HMBC correlations (Fig. 4) from 1-OH at [δ H 13.44 (s)] and 2-OCH3 at [δ H 3.72 (s)] to C-2 at [δ C 130.6 (s)]. All of these data for 6 were consistent with the structure 2,5-dimethoxy-8-methyl-1,3,6-trihydroxyxanthone.

Key HMBC () correlations of 6
It was worth mentioning that the 1H NMR data of 6 were identical to drimiopsin D (6a) isolated by D.A. Mulholland from Drimiopsis maculata [15]. However, there were some deviation in aspect of the 13C NMR data of 6 and 6a. We believed that the 13C NMR data of 6a should be measured in CD3OD. This inference was subsequently confirmed, in view of the complete consistency of the spectral data of 6 and 6a, when we re-tested its 13C NMR using CD3OD as deuterated solvent. As a conclusion, the structure of drimiopsin D (6a) was revised as 2,5-dimethoxy-8-methyl-1,3,6-trihydroxyxanthone (6) (Fig. 5).

Structure revision of drimiopsin D
3 Experimental
3.1 General Experimental Procedures
Optical rotations were measured on Jasco P-1020 or SGW®-3 (INESA Instrument Co., Ltd., Shanghai, China) automatic digital polarimeter. UV data were obtained from HPLC online analysis. IR spectra (KBr) were obtained on a Bruker Tensor-27 infrared spectrophotometer. NMR spectra were carried out on a Bruker Avance III 600 or Bruker AV-400 (Bruker BioSpin GmbH, Rheinstetten, Germany) spectrometer with deuterated solvent signals used as internal standards. ESIMS and HRESIMS were measured using Bruker HCT Esquire 3000 and API QSTAR time-of-flight mass spectrometers, respectively. Silica gel (200–300 mesh, Qingdao Marine Chemical Inc., China), MCI gel CHP-20P (75–150 μm, Mitsubishi Chemical Corporation, Japan), Chromatorex C-18 (40–75 μm, Fuji Silysia Chemical Ltd., Japan) and Sephadex LH-20 (Amersham Biosciences, Sweden) were used for column chromatography. Fractions were monitored and analyzed using TLC, in combination with Agilent 1200 series HPLC system equipped by Extend-C18 column (5 μm, 4.6 × 150 mm). A TCI Chiral MB-S column (5 μm, 4.6 × 250 mm, Tokyo Chemical Industry Co., Ltd., Tokyo, Japan) was applied for determination of acid hydrolyzates using Agilent 1200 series HPLC system with an external Alltech 3300 ELSD detector (Grace, Deerfield, USA).
3.2 Plant Material
The whole plants of S. scilloides were collected from He County of Anhui Province, China, in August 2009, and identified by Prof. Shou-Jin Liu of Anhui University of Chinese Medicine. A voucher specimen (No. BBP0208018SS) was deposited at BioBioPha Co., Ltd.
3.3 Extraction and Isolation
The dried and crushed whole plants of S. scilloides (9.5 kg) were extracted with EtOH-H2O (95:5, v/v; 3 × 18 L, each 7 days) at room temperature, and the solvent was removed under reduced pressure to give crude extract (ca. 1.9 kg), which was fractionated by silica gel CC successively eluted with a gradient of increasing acetone in petroleum ether (PE) (15:1, 10:1, 7:1, 4:1, 2:1, 1:1, 0:1; v/v) and then MeOH to afford fractions A–H, respectively. Fraction B was isolated by Sephadex LH-20 CC (CHCl3/MeOH; 1:1) and repeated silica gel CC (CHCl3/MeOH; 100:0 → 100:1) to yield 5 (181 mg). Fraction C was further separated by silica gel CC (CHCl3/MeOH; 100:1 → 20:1), Sephadex LH-20 (MeOH) and preparative TLC (PE/EtOAc; 8:1) to provide 2 (49 mg), 4 (474 mg) and 8 (45 mg). Fraction D was applied repeatedly to RP-18 CC (MeOH/H2O; 60 → 90 %) and then by Sephadex LH-20 (MeOH) to afford 1 (40 mg), 6 (127 mg) and 7 (116 mg). Compound 3 (291 mg) was eventually acquired by means of repeated silica gel CC (CHCl3/MeOH; 30:1 → 10:1), RP-18 (MeOH/H2O; 40 → 70 %) and Sephadex LH-20 (CHCl3/MeOH; 1:1) from the fraction F.
3.4 Scillascillol (1)
White amorphous powder; [α] 25D −0.7 [c 0.42, MeOH-CHCl3 (1:1)]; IR (KBr) νmax: 3441, 3432, 2943, 2880, 1765, 1631, 1455, 1374, 1321, 1116, 1036, 997, 944, 923 cm−1; 1H and 13C NMR data: see Table 1; ESIMS (pos.): m/z 525 [M + Na]+; HRESIMS (neg.): m/z 501.3207 [M − H]− (calcd for C30H45O6, 501.3216).
3.5 Scillascillone (2)
White amorphous powder; [α] 25D +46.0 (c 0.25, MeOH); IR (KBr) νmax: 3471, 2960, 2943, 1765, 1702, 1639, 1456, 1381, 1320, 1115, 1046, 998, 943, 923 cm−1; 1H and 13C NMR data: see Table 1; ESIMS (pos.): m/z 523 [M + Na]+; HRESIMS (neg.): m/z 499.3053 [M − H]− (calcd for C30H43O6, 499.3060).
3.6 Scillascilloside B-1 (3)
White amorphous powder; [α] 25D −57.9 (c 0.30, MeOH); IR (KBr) νmax: 3440, 2940, 2883, 1774, 1725, 1713, 1633, 1456, 1376, 1255, 1078, 1048, 1010, 755 cm−1; 1H and 13C NMR data: see Table 2; ESIMS (pos.): m/z 775 [M + Na]+; HRESIMS (neg.): m/z 751.4261 [M − H]− (calcd for C40H63O13, 751.4269).
3.7 2,5-Dimethoxy-8-methyl-1,3,6-trihydroxyxanthone (= drimiopsin D, 6)
Yellow amorphous powder; UV (MeOH) λmax: 247, 276 (sh), 321 nm; IR (KBr) νmax: 3443, 2959, 2849, 1650, 1619, 1579, 1513, 1461, 1320, 1292, 1267, 1186, 1162, 1105, 992, 816, 796 cm−1; 1H and 13C NMR data: see Table 3; ESIMS (pos.): m/z 341 [M + Na]+; HRESIMS (neg.): m/z 317.0665 [M − H]− (calcd for C16H13O7, 317.0661).
3.8 Acid Hydrolysis of 3
Compound 3 (3 mg) was heated in 2 M HCl (3 mL) at a temperature of 45 °C for 4 h. After cooling, the reaction mixture was neutralized with KOH to a pH of approximately 7, and then extracted with EtOAc. The aqueous layer was analyzed by HPLC under the following conditions: solvent, n-hexane/isopropanol (85:15); flow rate, 1.0 ml/min; temperature, 25 °C. The retention times (t R) of d-glucose and l-arabinose were 5.5 and 6.9 min, respectively.
References
M. Pfosser, F. Speta, Ann. Missouri Bot. Gard. 86, 852–875 (1999)
M.Y. Fan, Y.M. Wang, Z.M. Wang, H.M. Gao, Chin. J. Chin. Mater. Med. 39, 162–170 (2014)
I. Kouno, N. Noda, Y. Ida, M. Sholichin, K. Miyahara, T. Komori, T. Kawasaki, Liebigs Ann. Chem. 1982, 306–314 (1982)
M. Sholichin, K. Miyahara, T. Kawasaki, Heterocycles 17, 251–257 (1982)
M. Sholichin, K. Miyahara, T. Kawasaki, Chem. Pharm. Bull. 33, 1756–1759 (1985)
S.M. Lee, H.K. Chun, C.H. Lee, B.S. Min, E.S. Lee, Y.H. Kho, Chem. Pharm. Bull. 50, 1245–1249 (2002)
Y. Nishida, M. Eto, H. Miyashita, T. Ikeda, K. Yamaguchi, H. Yoshimitsu, T. Nohara, M. Ono, Chem. Pharm. Bull. 56, 1022–1025 (2008)
M. Ono, D. Toyohisa, T. Morishita, H. Horita, S. Yasuda, Y. Nishida, T. Tanaka, M. Okawa, J. Kinjo, H. Yoshimitsu, T. Nohara, Chem. Pharm. Bull. 59, 1348–1354 (2011)
M. Ono, Y. Takatsu, T. Ochiai, S. Yasuda, Y. Nishida, T. Tanaka, M. Okawa, J. Kinjo, H. Yoshimitsu, T. Nohara, Chem. Pharm. Bull. 60, 1314–1319 (2012)
M. Ono, T. Ochiai, S. Yasuda, Y. Nishida, T. Tanaka, M. Okawa, J. Kinjo, H. Yoshimitsu, T. Nohara, Chem. Pharm. Bull. 61, 592–598 (2013)
F. Wang, Y. Gao, L. Zhang, B. Bai, Y.N. Hu, Z.J. Dong, Q.W. Zhai, H.J. Zhu, J.K. Liu, Org. Lett. 12, 3196–3199 (2010)
F. Wang, Y.J. Li, F.C. Ren, G.Z. Wei, J.K. Liu, Chem. Pharm. Bull. 59, 484–487 (2011)
F. Wang, D.S. Zhou, G.Z. Wei, F.C. Ren, J.K. Liu, Phytochemistry 77, 312–317 (2012)
F. Wang, X.L. Li, G.Z. Wei, F.C. Ren, J.K. Liu, Nat. Prod. Bioprospect. 3, 238–242 (2013)
D.A. Mulholland, C. Koorbanally, N.R. Crouch, P. Sandor, J. Nat. Prod. 67, 1726–1728 (2004)
A. Abdel-Lateff, C. Klemke, G.M. König, A.D. Wright, J. Nat. Prod. 66, 706–708 (2003)
Y. Mimaki, S. Kubo, Y. Kinoshita, Y. Sashida, L.G. Song, T. Nikaido, T. Ohmoto, Phytochemistry 34, 791–797 (1993)
K. Ori, M. Koroda, Y. Mimaki, H. Sakagami, Y. Sashida, Phytochemistry 64, 1351–1359 (2003)
H.T. Nguyen, G.Y. Song, J.A. Kim, J.H. Hyun, H.K. Kang, Y.H. Kim, Bioorg. Med. Chem. Lett. 20, 309–314 (2010)
D.C. Albach, C.H. Gotfredsen, S.R. Jensen, Phytochemistry 65, 2129–2134 (2004)
T. Kanchanapoom, Phytochemistry 68, 692–696 (2007)
Y.B. Zhuang, H. Yin, X.W. Zhang, W. Zhou, T. Liu, Helv. Chim. Acta 98, 699–703 (2015)
Acknowledgments
This work was financially supported by “Large-scale Compound Library” project of National Development and Reform Commission of China.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ren, FC., Wang, LX., Yu, Q. et al. Lanostane-Type Triterpenoids from Scilla scilloides and Structure Revision of Drimiopsin D. Nat. Prod. Bioprospect. 5, 263–270 (2015). https://doi.org/10.1007/s13659-015-0076-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-015-0076-0