
Abstract
Investigation on the cultures of Hypsizygus marmoreus resulted in the isolation of a new viscidane diterpene, 8-oxoviscida-3,11(18)-diene-13,14,15,19-tetraol (1) and two new polyacetylenes, (E)-10-(1,1-dimethyl-2-propenyloxy)-2-decene-4,6,8-triyn-1-ol (2) and 10-(1,1-dimethyl-2-propenyloxy)deca-4,6,8-triyn-1-ol (3), together with two known polyacetylenes, (E)-2-decen-4,6,8-triyn-1-ol (4) and 4,6,8-decatriyn-1-ol (5). Their structures were elucidated on the basis of extensive spectroscopic studies. Compound 1 is the first finding of viscidane diterpene in mushrooms. Compounds 1, 3 and 5 were tested for cytotoxicity against human tumor cell lines HL-60, SMMC-7721, A-549, MCF-7 and SW-480. None of the compounds showed cytotoxic activity (IC50 > 40 µM).
Graphical Abstract

Similar content being viewed by others
1 Introduction
The fungus Hypsizygus marmoreus (Tricholomataceae) is a group of edible mushrooms native to East Asia, occurring in autumn and winter of the north temperature zone. In a previous investigation of their fruiting bodies led to the isolation of steroids [1, 2], sphingolipids [3], proteins [4], and polyisoprenepolyols [5–7]. However, the cultures of this mushroom have not been chemically investigated so far. In continuation of our study of the secondary metabolites from the untapped resources of higher fungi collected in China [8–11], we have investigated the cultures of H. marmoreus, which led to the isolation of three new compounds, a viscidane diterpene (1) and two polyacetylenes (2 and 3), as well as two known polyacetylenes (4 and 5). Viscidane diterpenes are characteristic constituents in Eremophila species, nevertheless, 1 is the first example of this type found in higher fungi. Furthermore, polyacetylenes usually possess high antifungal and nematicidal activities and are phototoxic against certain viruses [12–16]. In macrofungi, the incorporation of oleic, linoleic, crepenynic, and dehydrocrepenynate into polyacetylenes is well established [17]. The present paper reports the isolation and structure determination of the new compounds.
2 Results and Discussion
An EtOAc extracts (4.0 g) of the culture broth (18 L) of H. marmoreus was subjected to silica gel column chromatography (CC) with a gradient elution system of petroleum ether–acetone (100:0–0:100) to obtain eight fractions. Fractions 3, 5 and 7 were further chromatographed on Sephadex LH-20 CC (CHCl3–MeOH, 1:1) and purified by preparative HPLC (Pre-HPLC, MeCN–H2O) to give three new compounds 1–3 and known ones. The known compounds were determined to be (E)-2-decen-4,6,8-triyn-1-ol (4) [18] and 4,6,8-decatriyn-1-ol (5) [19].
Compound 1, obtained as oil, had the molecular formula C20H32O5 based on the HRESIMS (pos.), showing a quasi-molecular ion peak at m/z 375.2147 (calcd for C20H32O5Na, 375.2147) with five degrees of unsaturation. In accordance with the molecular formula, 20 carbon resonances were resolved in the 13C NMR spectrum (Table 1), including a saturated ketone at δ C 222.0 (s), a set of signals at δ C 149.0 (s), 134.3 (s), 121.4 (d), 114.6 (t) assignable to a terminal double bond and a trisubstituted one, three oxygen-bearing carbons at δ C 78.7 (d), 74.6 (s) and 70.2 (d), as well as three methyl signals at δ C 27.2 (q), 26.4 (q) and 23.5 (q). The 1H NMR spectrum (Table 1), in combination with the HSQC spectrum, exhibited three vinyl signals at δ H 5.35 (1H, br. s), 5.08 (1H, br. s) and 4.77 (1H, br. s), four oxygen-bearing protons at δ H 4.01 (1H, br. dd, J = 8.7, 4.7 Hz), 3.83 (1H, dd, J = 11.4, 5.3 Hz), 3.71 (1H, dd, J = 11.4, 5.8 Hz) and 3.06 (1H, br. s), three spin-coupled protons at δ H 3.05 (1H, dd, J = 8.8, 4.4 Hz), 2.53 (1H, dd, J = 19.4, 8.8 Hz) and 2.41 (1H, dd, J = 19.4, 4.4 Hz), and three tertiary methyls signals at δ H 1.67 (3H, s), 1.24 (3H, s), and 1.22 (3H, s). According to the degrees of unsaturation, this molecule contained two rings. The above NMR character was very similar with that of a known compound viscida-3,11(18),14-triene [20]. Nevertheless there were obvious differences: the absence of a trisubstituted double bond signals, a doublet methyl signals and two high field methylene signals, instead a saturated carbonyl carbon signal and four oxygen-bearing carbon signals were observed. The carbonyl and hydroxymethyl were posited at C-8 and C-7 respectively, as established by the HMBC correlations (Table 1) of δ H 3.83 (1H, dd, J = 11.4, 5.3 Hz, H-19a) and 3.71 (1H, dd, J = 11.4, 5.8 Hz, H-19b) with δ C 45.1 (s, C-1), 58.7 (d, C-7) and 222.0 (s, C-8); of δ H 2.45 (1H, dd, J = 5.8, 5.3 Hz, H-7), 2.53 (1H, dd, J = 19.4, 8.8 Hz, H-9a), 2.41 (1H, m, H-9b), 3.05 (1H, m, H-10), 3.83 (1H, dd, J = 11.4, 5.3 Hz, H-19a) and 3.71 (1H, dd, J = 11.4, 5.8 Hz, H-19b) with δ C 222.0 (s, C-8). HMBC correlations also evidenced that C-13, C-14 and C-15 in 1 were hydroxylated. From ROESY experiment (Fig. 2), the significant correlations of H-10/H-19 and H-19/H-6 were observed, indicating α-orientation of these protons. In combination with the comparison of relevant NMR chemical shifts and 1H–1H coupling constants with those of analogues [21, 22], the configuration of 1 was established as shown in Fig. 1. Accordingly, the structure of 1 was determined and named as 8-oxoviscida-3,11(18)-diene-13,14,15,19-tetraol.

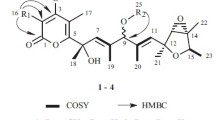
Structures of compounds 1–5

Key ROESY correlations of 1
Compound 2 was isolated as yellow oil. HREIMS analysis gave the molecular formula C15H16O2 (m/z 228.1158, calcd 228.1150), requiring eight degrees of unsaturation. The compound showed a UV spectrum typical of a triyne at 331, 309, 291, 274, 258 and 244 nm [19], whereas its IR spectrum indicated the presence of hydroxy (3406 cm−1), triyn (2179 cm−1) and acetylene (1626, 946 cm−1). The 1H NMR spectrum (Table 1) showed resonances for a terminal double bond at δ H 5.80 (1H, dd, J = 17.4, 11.1 Hz, H-2′), 5.18 (1H, br. d, J = 11.1 Hz, H-3′) and 5.17 (1H, br. d, J = 17.4 Hz, H-3′), a trans disubstituted double bond at δ H 6.48 (1H, dt, J = 15.9, 4.5 Hz, H-2) and 5.83 (1H, d, J = 15.9 Hz, H-3), two oxygenated methylene protons at δ H 4.27 (2H, d, J = 4.5 Hz, H-1) and 4.09 (2H, s, H-10), and two methyl signals at δ H 1.31 (6H, s, H-4′ and H-5′). In combination with HMQC spectrum, 15 protons were unambiguously assigned to their corresponding carbon atoms. However, according to the molecular formula, one proton signal was not observed, which can presumably be attributed to a hydroxy group. The 13C NMR (DEPT) spectrum (Table 2) exhibited 15 carbon signals, consisting of two methyls, three methylenes (including two oxidized carbons), three olefinic methines, and seven quaternary carbons (including a triyn group). The 1H and 13C NMR data of 2 were similar to those of (E)-2-decen-4,6,8-triyn-1-ol (4) [18], except for the additional signals at δ C 142.5 (d, C-2′), 115.2 (t, C-3′), 77.1 (s, C-1′), 25.7 (q, C-4′ and C-5′) and 51.9 (t, C-10), and the absence of the methyl signal at δ C 4.6 (q, C-10), indicating that the methyl at C-10 in 4 was replaced by a reverse isoprene 2-methylbut-3-en-2-yloxy moiety in 2. This conclusion was supported by the HMBC correlations from 5.18 (1H, br. d, J = 11.1 Hz, H-3′) and 5.17 (1H, br. d, J = 17.4 Hz, H-3′) to 142.5 (d, C-2′), 77.1 (s, C-1′) and 25.7 (q, C-4′ and C-5′), from 4.09 (2H, s, H-10) to 77.1 (s, C-1′), and from 1.31 (6H, s, H-4′ and H-5′) to 142.5 (d, C-2′) and 77.1 (s, C-1′). The C-2/C-3 olefin was assigned the E-geometry based on the characteristic vicinal coupling constant of H-2 and H-3 (J = 15.9 Hz). Thus, the structure of 2 was determined as (E)-10-(1,1-dimethyl-2-propenyloxy)-2-decene-4,6,8-triyn-1-ol.
Compound 3 was also obtained as yellow oil. Its molecular formula was determined as C15H18O2 from the HREIMS ion peak at 230.1294 (C15H18O2, calcd 230.1307), representing an unsaturation value of seven. Comparing the NMR data of 3 (Table 2) with those of 2, 3 showed evidence of having two more methylenes (δ C 30.6 and 15.9) and two less olefinic methines (δ C 147.3 and 108.1) than 1. Further evidence from the HMBC spectrum of 3, in which the proton at 2.43 (2H, t, J = 7.0 Hz, H-3) correlated to C-1 (61.2, t) and 30.6 (t, C-2), and the proton at 1.78 (2H, q-like, H-2) correlated to C-4 (79.9, s), C-1 (61.2, t), C-2 (30.6, t) and C-3 (15.9, t), suggesting that a double bond between C-2 and C-3 was hydrogenated in 3. Therefore, compound 3 was assigned as 10-(1,1-dimethyl-2-propenyloxy)deca-4,6,8-triyn-1-ol.
3 Experimental
3.1 General Experimental Procedures
Optical rotations were measured on a Jasco P-1020 (Jasco International Co., Ltd., Tokyo, Japan) automatic digital polarimeter. IR spectra were recorded using a Bruker Tensor 27 FT-IR (Bruker Optics GmbH, Ettlingen, Germany) spectrometer with KBr pellets. UV spectra were carried out on a Shimadzu UV-2401A spectrometer. NMR spectra were carried out on Bruker DRX-500, AV-400 or AV-600 (Bruker BioSpin GmbH, Rheinstetten, Germany) spectrometer with the deuterated solvent as an internal standard. EIMS and ESIMS (including HRESIMS) were measured on Finnigan-MAT 90 and API QSTAR Pulsar i (MDS Sciex, Concord, Ontario, Canada) mass spectrometers, respectively. Silica gel 200–300 mesh (Qingdao Marine Chemical Inc., Qingdao, China) and Sephadex LH-20 (Amersham Biosciences, Uppsala, Sweden) were used for normal pressure column chromatography. Fractions were monitored and analyzed by TLC (silica gel 60 F254, Qingdao Marine Chemical Inc., Qingdao, China), in combination with Agilent 1200 series HPLC system (Eclipse XDB-C18 column, 5 μm, 4.6 × 150 mm). Preparative HPLC was performed using an Agilent 1100 series (Zorbax SB-C18 column, 5 μm, 9.4 × 150 mm).
3.2 Fungal Material and Cultivation Conditions
The fungus H. marmoreus was provided by Huazhong Agricultural University (cultivated), in 2000, and identified by Prof. Yu-Cheng Dai, Beijing Forestry University. The voucher specimen (MC00110) was deposited at the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, CAS. Culture PDA medium: potato (peeled), 200 g, glucose, 20 g, KH2PO4, 3 g, MgSO4, 1.5 g, citric acid, 0.1 g, and thiamin hydrochloride, 10 mg, in 1 L of deionized H2O. Reagent bottles were used as a flask (size: 500 mL; volume of media: 300 mL). The pH was adjusted to 6.5 before autoclaving, and the fermentation was carried out on a shaker at 25 and 150 rpm for 30 days.
3.3 Extraction and Isolation
The culture broth (18 L) was extracted three times with EtOAc (36 L). The combined EtOAc extracts were evaporated in vacuo to give a residue (4.0 g). The residue was subjected to silica gel column chromatography (CC) with a gradient elution system of petroleum ether–acetone (100:0–0:100) to obtain eight fractions. Fraction 3 was eluted with petroleum ether–acetone (10:1). It was then subjected to Sephadex LH-20 CC (CHCl3–MeOH, 1:1) and Pre-HPLC (35–50 % MeCN in H2O over 40 min, 10 mL/min) to yield 2 (4.2 mg), 3 (12.5 mg), 4 (12.0 mg) and 5 (10.3 mg). The fraction 7 (petroleum ether–acetone, 3:1) was further chromatographed on Sephadex LH-20 CC (CHCl3–MeOH, 1:1) and then purified by Pre-HPLC (4–30 % MeCN in H2O over 40 min, 10 mL/min) to afford 1 (2.4 mg).
3.4 8-Oxoviscida-3,11(18)-diene-13,14,15,19-tetraol (1)
Oil, [α] 10D –3.9 (c 0.16, CH3OH). 1H and 13C NMR: see Table 1. ESIMS (pos.): 375 [M + Na]+. HRESIMS (pos.): 375.2147 (C20H32O5Na, calcd 375.2147).
3.5 (E)-10-(1,1-Dimethyl-2-propenyloxy)-2-decene-4,6,8-triyn-1-ol (2)
Yellow oil. UV λ max (MeOH): 331, 309, 291, 274, 258, 244 nm; IR (KBr): 3406, 3087, 2977, 2926, 2852, 2179, 1626, 946, 935 cm−1. 1H and 13C NMR: see Table 2. EIMS: 228 [M]+. HREIMS: 228.1158 (C15H16O2, calcd 228.1150).
3.6 10-(1,1-Dimethyl-2-propenyloxy)deca-4,6,8-triyn-1-ol (3)
Yellow oil. UV λ max (MeOH): 283, 266, 253, 213, 204 nm; IR (KBr): 3405, 3086, 2978, 2933, 2854, 2218, 1718, 1626, 1145, 1057, 930 cm−1. 1H and 13C NMR: see Table 2. EIMS: 230 [M]+. HREIMS: 230.1294 (C15H18O2, calcd 230.1307).
3.7 Cytotoxicity Assay
The following human tumor cell lines were used: HL-60, SMMC-7721, A-549, MCF-7 and SW-480. All the cells were cultured in RPMI-1640 or Dulbecco’s modified Eagle’s medium (DMEM) (Hyclone, USA), supplemented with 10 % fetal bovine serum (Hyclone, USA) at 37 °C in a humidified atmosphere with 5 % CO2. Cell viability was assessed by conducting colorimetric measurements of the amount of insoluble formazan that formed in living cells based on the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma, USA) [23]. Briefly, 100 mL of adherent cells were seeded into each well of 96-well cell culture plates and allowed to adhere for 12 h before drug addition, while suspended cells were seeded just before drug addition, both with an initial density of 1 × 105 cells/mL in 100 μL of medium. Each tumor cell line was exposed to the tested compound at various concentrations in triplicate for 48 h, with 10-hydroxy-campto-thecine (Sigma, USA) as positive control. After the incubation, MTT (100 mg) was added to each well, and the incubation continued for 4 h at 37 °C. The cells were lysed with 100 μL of 20 % SDS–50 % DMF after removal of 100 μL of medium. The optical density of the lysate was measured at 595 nm in a 96-well microtiter plate reader (Bio-Rad 680, USA). The IC50 value of each compound was calculated by the Reed and Muench method [24].
References
Y. Yaoita, K. Amemiya, H. Ohnuma, K. Furumura, A. Masaki, T. Matsuki, M. Kikuchi, Chem. Pharm. Bull. 46, 944–950 (1998)
Y. Yaoita, Y. Endo, Y. Tani, K. Machida, K. Amemiya, K. Furumura, M. Kikuchi, Chem. Pharm. Bull. 47, 847–851 (1999)
S. Akiyoshi, M. Masanori, O. Seiji, O. Tadashi, Advances in Mass Spectrometry (Elsevier Science, Amsterdam, 1998), p. 14
S. Akiyoshi, M. Masanori, O. Seiji, O. Tadashi, J. Mass Spectrom. 31, 921–925 (1996)
S. Akiyoshi, M. Masanori, K. Tatsuya, K. Hideki, O. Yoshikazu, O. Seiji, O. Tadashi, J. Agric. Food Chem. 47, 588–593 (1999)
Y. Shoji, M. Isemura, H. Muto, S. Isemura, Y. Aoyagi, Biosci. Biotechnol. Biochem. 64, 775–780 (2000)
S. Akiyoshi, M. Masanori, O. Seiji, O. Tadashi, Kinki Daigaku Nogaku Sogo Kenkyusho Hokoku 9, 97–105 (2001)
J.K. Liu, Chem. Rev. 106, 2209–2223 (2006)
J.K. Liu, Chem. Rev. 105, 2723–2744 (2005)
D.Z. Liu, F. Wang, J.K. Liu, J. Nat. Prod. 70, 1503–1506 (2007)
M.Y. Jiang, L. Zhang, R. Liu, Z.J. Dong, J.K. Liu, J. Nat. Prod. 72, 1405–1409 (2009)
V.M. Dembitsky, D.O. Levitsky, Nat. Prod. Commun. 1, 405–429 (2006)
V.M. Dembitsky, Lipids 41, 883–924 (2006)
L.P. Christensen, K. Brandt, Pharm. Biomed. Anal. 41, 683–693 (2006)
R. Ebermann, G. Alth, M. Kreitner, A. Kubin, J. Photochem. Photobiol. B 36, 95–97 (1996)
M. Heinrich, M. Robles, J.E. West, B.R. Ortiz de Montellano, E. Rodriguez, Annu. Rev. Pharmacol. Toxicol. 38, 539–565 (1998)
R.E. Minto, B.J. Blacklock, Prog. Lipid Res. 47, 233–306 (2008)
M.T.W. Hearn, J.L. Turner, J. Chem. Soc. Perkin Trans. 2, 1027–1029 (1976)
A.A. Taha, Phytochemistry 55, 921–926 (2000)
H. Tesso, W.A. König, Y. Asakawa, Phytochemistry 66, 941–949 (2005)
E.L. Glzisalberti, C.H. Hocart, P.R. Jefiries, G.M. Proudjoot, B.W. Skelton, A.H. White, Aust. J. Chem. 36, 993–1000 (1983)
E.L. Ghisalberti, P.R. Jefferies, T.A. Mori, Aust. J. Chem. 37, 635–647 (1984)
T. Mosmann, J. Immunol. Methods 65, 55–63 (1983)
L.J. Reed, H. Muench, Am. J. Hyg. 27, 493–497 (1938)
Acknowledgments
This project was supported by the National Natural Sciences Foundation of China (21302194).
Conflict of Interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Zhang, L., Li, ZH., Dong, ZJ. et al. A Viscidane Diterpene and Polyacetylenes from Cultures of Hypsizygus marmoreus . Nat. Prod. Bioprospect. 5, 99–103 (2015). https://doi.org/10.1007/s13659-015-0058-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-015-0058-2