
Abstract
A new limonoid, 17-(5-methoxy-2-oxofuran-3-yl)-28-deoxonimbolide (1), and a new C21 steroidal saponin, 2α,4α-dihydroxy-pregn-5-en-16-one-3α-O-d-glucopyranoside (2), together with 11 known compounds were isolated from the methanol extract of the leaves of Azadirachta indica. The structures were elucidated by means of spectroscopic analysis and putative biosynthetic origins. All the compounds were evaluated for their antibacterial activities against six bacterial strains.
Graphical Abstract

Similar content being viewed by others


1 Introduction
Herbal medicines are widely used and formed as an integral part of primary health care in many countries [1–3], and some are used to treat fungal diseases, which may constitute a reservoir of antifungal substances. In recent years, a number of antifungal agents are currently used in antifungal therapies with clinical practice, such as griseofulvin and terbinafine [4, 5]. These antifungals are isolated from fungi or are synthetized, and searching for new antifungal substances from plants is still challenging [6].
Azadirachta indica (neem tree), indigenous to India, belongs to the Meliaceae family, are widely cultivated throughout the tropics and subtropics [7]. In view of therapeutic and bioactive importance of the plant, it has attracted the attention of investigators all over the world to research bioactive compounds [8–10]. Antibacterial effects of neem extract have been demonstrated against some Gram-negative and Gram-positive microorganisms, including Streptococcus mutans, S. faecalis, Escherichia coli and Pseudomonas florescence pathogenic strains [11, 12]. In a continuation of our studies on its constituents [8–10], a new limonoid, 17-(5-methoxy-2-oxofuran-3-yl)-28-deoxonimbolide (1) and a new C21 steroid saponin, 2α,4α-dihydroxy-pregn-5-en-16-one-3α-O-d-glucopyranose (2), together with 6-deacetylnimbin (3) [13], 6-deacetylnimbinal (4) [13], nimbandiol (5) [14], nimbolide (6) [13], 2′,3′-dehydrosalannol (7) [15], 3β,4β,20α-trihydroxy-5-pregnen (8) [16], 2α,3β-dihydroxy-5-pregnen-16-one (9) [17], (+)-dehydro-vomifoliol (10) [18], 3β-hydroxy-5α,6α-epoxy-7-megastigmen-9-one (11) [19], quercetin-3-glucopyranside (12) [20] and quercetin-3-glu (6→1) rha (13) [21] were isolated from the methanol extract of the leaves of A. indica (Fig. 1). The structures were elucidated by means of spectroscopic analysis. All the compounds were evaluated for their antibacterial activities against six bacterial strains.

Chemical structures of compounds 1–13
2 Results and discussion
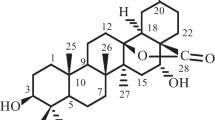
Compound 1 exhibited a molecular formula of C28H34O8 by HR-EI-MS (m/z 498.2246 [M]+), indicating 12 degrees of unsaturation. The UV maximum at 240 nm indicated the existence of α,β-unsaturated ketone system, while the IR spectrum showed the absorption bands of γ-lactone (1759 cm−1), ester C=O (1728 cm−1), and conjugated cyclohexenone (1631 cm−1) chromophoric groups. Its 1H NMR spectrum showed three olefinic proton signals at δH 7.04 (d, J = 9.7 Hz), 5.86 (d, J = 9.7 Hz) and 6.82 (s), four methyl signals at δH 1.72 (s), 1.14 (s), 1.33 (s) and 1.29 (s), two methoxyl signals at δH 3.63 (s) and 3.50 (s).
The 13C NMR and DEPT spectra of 1 displayed 28 carbons signals comprising two ester carbonyl groups, one ketone carbonyl group, three double bonds, three quaternary carbons, seven methines (four oxygenated ones), three methylenes (one oxygenated carbon), six methyls (Table 1). These aforementioned data suggested that compound 1 is a nimbolide-type limonoid, structurally similar to 28-deoxonimbolide [13]. The α,β-unsaturated-γ-lactone moiety was suggested on the basis of four carbons at δC 139.4 (s, C-20), 171.3 (s, C-21), 142.3 (d, C-22), and 102.3 (d, C-23), and the corresponding protons at δH 6.82 (s, H-22) and 5.73 (s, H-23). The assumption was supported by the 1H-1H COSY spin system of H-22/H-23, and by the HMBC correlations of δH 6.82 (s, H-22) and 5.73 (s, H-23) with δC 171.3 (s, C-21) and 139.4 (s, C-20). In addition, the HMBC correlation of methoxyl protons at δH 3.50 with a hemiketal group at δC 102.3 (d, C-23), suggested the methoxyl group was placed at C-23. In the ROESY spectrum, correlations of Me-29 with H-6, of Me-30 with H-7 positioned β-orientation for both H-6 and H-7. The ROESY correlations of H-5/H-9/H-15, indicated the α-orientation of H-15. Other parts of 1 were identical to those of 28-deoxonimbolide, as further confirmed by detailed analysis of 2D NMR spectra of 1. Thus, the structure of 1 was elucidated as shown (Fig. 1), and named as 17-(5-methoxy-2-oxofuran-3-yl)-28-deoxonimbolide.
Compound 2 was assigned a molecular formula of C27H42O9, by its HR-EI-MS peak at m/z 510.2833 ([M]+ calcd. for 510.2829), in combination with 1H, 13C NMR and DEPT data, corresponding to 7 degrees of unsaturation. IR absorptions bands at 3428 and 1631 cm−1 revealed the existence of OH and C=C groups. Signals of two high-field quaternary carbons (δC 38.5, 42.9) in the 13C NMR spectrum, along with two singlet methyls (δH 0.74, 1.29) and a triplet methyl (δH 1.03) (Table 1) in the 1H NMR spectrum, suggested that compound 2 was a pregnane derivative [15]. Correlations of δH 1.77 (H-17) with δC 221.8 (s, C-16), and of δH 1.54 (H-14) with δC 221.8 (s, C-16) in the HMBC spectrum assigned C-16 as a ketone (Fig. 2). The HMBC correlations of δH 4.37 (d, J = 3.5 Hz, H-4)/δC 142.4 (s, C-5), of δH 1.29 (s, Me-19)/δC 142.4 (s, C-5), and of δH 5.76 (t, J = 3.7 Hz, H-6)/δC 38.5 (s, C-10) suggested the existence of double bond between at C-5 and C-6. The 1H-, 13C-NMR, and DEPT spectra displayed a d-glucopyranosyl unit on the basis of an anomeric proton signal at δH 4.41 (d, J = 7.7 Hz, H-1′), an methylene signals at δH 3.64 (m, H-6′) and 3.84 d (m, H-6′), four additional protons between δH 3.29 and δH 3.38; as well as an anomeric C-atom (δC 102.5, d), an methylene group (δC 66.9, t), and other four CH groups [δC 74.9 (d), 77.6 (d), 71.5 (d), 78.1 (d)]. The sugar unit was further verified as d-glucopyranose by GC analysis of its corresponding trimethylsilylated l-cysteine adduct after acidic hydrolysis of 2 [22]. Correlations of δH 3.44 (dd, J = 3.5, 3.6 Hz, H-3)/δC 102.5 (d, C-1′), and of δH 4.41 (d, J = 7.7 Hz, H-1′)/δC 85.5 (d, C-3) in the HMBC spectrum indicated the sugar unit was linked at C-3. In the ROESY spectrum, correlations of δH 3.44 (Me-19)/4.11 (H-2)/4.37 (H-4), and of δH 3.44 (Me-19)/1.09 (H-1a)/3.44 (H-3) positioned β-orientation for H-2, H-3 and H-4, and α-orientation for OH-2, O-Glc-3 and OH-4 (Fig. 2). Thus, the structure of 2 was elucidated as shown, and named as 2α,4α-dihydroxy-pregn-5-en-16-one-3α-O-d-glu-copyranoside.

Selected COSY ( ), HMBC (
), HMBC ( ) and ROESY (
) and ROESY ( ) correlations of compound 2
) correlations of compound 2
All the compounds were evaluated for their antibacterial activities against Escherichia coli ATCC 11775, Enterococcus faecalis ATCC 10541, Klebsiella pneumonia ATCC 13883, Pseudomonas aeruginosa ATCC 27853, Staphylococcus aureus ATCC 25922, and Salmonella enterica ATCC 13076. Norfloxacin was used as the positive control. The results showed that compounds 1 and 2 exhibited strong antibacterial activities against some bacterial strains, equivalent to norfloxacin with MIC values of 0.78 μg/mL (Table 2). And the other results (MIC values) are summarized in Table 2.
3 Experimental Section
3.1 General Experimental Procedures
Optical rotations were obtained with a Jasco P-1020 Automatic Digital Polariscope. UV spectrum was measured with a Shi madzu UV2401PC spectrometer. IR spectra were obtained on a Bruker FT-IR Tensor-27 infrared spectrophotometer with KBr pellets. 1H, 13C, and 2D NMR spectra were recorded on a Bruker AM-400, Bruker DRX-500 NMR and Bruker DRX-600 spectrometer with TMS as internal standard. ESI-MS and HR-EI-MS analysis were carried out on Waters Xevo TQS and Waters AutoSpec Premier P776 mass spectrometers, respectively. Semi-preparative HPLC was performed on an Agilent 1100 HPLC with a ZORBAX SB-C18 (9.4 × 250 mm) column. Silica gel (100–200 and 200–300 mesh, Qingdao Marine Chemical Co. Ltd., P.R. China), Sephadex LH-20 (GE Healthcare Bio-Xciences AB), and MCI gel (75–150 μm, Mitsubishi Chemical Corporation, Tokyo, Japan) were used for column chromatography. Fractions were monitored by TLC (GF 254, Qingdao Marine Chemical Co. Ltd., P.R. China), and spots were visualized by 10 % H2SO4-ethanol reagent.
3.2 Plant Material
The leaves of A. indica were collected from Xishuangbanna county, Yunnan province, China, in June 2012, and were identified by Dr. Jian Liu. A voucher specimen (Luo20120620) has been deposited at the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences.
3.3 Extraction and Isolation
The leaves of A. indica A. Juss. (10 kg) were powdered and extracted with MeOH (30 L percolation, 12 h × 3) to yield a MeOH extract (1745 g). The MeOH extract was suspended in H2O (4 L) and partitioned successively with EtOAc (3 L × 3) to yield an EtOAc fraction (320 g) and a H2O-soluble fraction (1425 g). The EtOAc fraction was subjected to silica gel column chromatography eluted with a gradient solvent system of chloroform/acetone (from 1:0 to 0:1, v/v) to afford five fractions (A–E) on the basis of TLC detection. First of all, fraction D (200 g, MeOH/H2O) was chromatographed on MCI column for removing the color, and then by RP-18 column chromatography (MeOH–H2O) to yield subfractions a–c. Each subfraction was purified by RP C-18 (MeOH–H2O) silica gel column (chloroform/acetone system), preparative thick layer chromatography (PTLC), and Sephadex LH-20 column (MeOH) successively, to yield compounds 1 (15 mg), 3 (950 mg), 4 (500 mg), 5 (4.1 g), 6 (560 mg), and 7 (6.8 g). Fraction E (93 g) was subjected to MCI column, silica gel column chromatography, PTLC, and Sephadex LH-20 column in turns to give compounds 2 (8 mg), 8 (4.1 g), 9 (15 mg), 10 (20 mg), 11 (13 mg), 12 (50 mg) and 13 (70 mg).
3.4 17-(5-Methoxy-2-oxofuran-3-yl)-28-deoxonimbolide (1)
White powder; [α] 21D +112.3 (c 0.11, CHCl3); UV (CHCl3) λmax (log ε) 240.8 (3.63); IR (KBr) νmax 3428, 2933, 2877, 2854, 1759, 1728, 1631, 1384 cm−1; 1H and 13C NMR data see Table 1; positive ESIMS m/z 521 [M+Na]+; HREIMS m/z 498.2246 (calcd for C28H34O8 [M]+, 498.2254).
3.5 2α,4α-Dihydroxy-pregn-5-en-16-one-3α-O-d-glucopyranoside (2)
White powder; [α] 21D −112.1 (c 0.10, MeOH); IR (KBr) νmax 3428, 2933, 2877, 2854, 1738, 1631, 1384 cm−1; 1H and 13C NMR data see Table 1; positive ESIMS m/z 533 [M+Na]+; HREIMS m/z 510.2833 (calcd for C27H42O9 [M]+, 510.2829).
3.6 Acidic Hydrolysis of 2 and GC analysis
Compound 2 (2 mg) was hydrolyzed with 2 M HCl/dioxane (1:1, 10 mL) under reflux for 3 h. The reaction mixture was partitioned between CHCl3 and H2O. The aqueous layer was neutralized with 2 M NaOH and then dried to give a sugar. The sugar was dissolved in anhydrous pyridineand (1 mL) and reacted with l-cysteine methyl ester hydrochloride (1.5 mg) stirred at 60 °C for 1.5 h. Then trimethylsilylimidazole (1.0 mL) was added to the reaction mixture, and it was kept at 60 °C for 30 min. The mixture (4 μL) was subjected to GC analysis, run on an HP 5890 gas chromatograph (Agilent) with a quartz capillary column (30 mm × 0.32 mm × 0.25 μm): H2 flame ionization detector, column temp 180–280 °C at 3 °C/min, carrier gas N2 (1 mL/min), injector and detector temp 250 °C, split ratio 1:50. Peak of the hydrolysate was detected by comparison with retention time of authentic samples of d-glucose after treatment with trimethyl-chlorsilan (TMCS) in pyridine. The absolute configurations of the sugar residue was determined to be d-glucose (tR 19.01 min).
3.7 Antimicrobial Assays
The antibacterial assay of compounds 1–13 was evaluated against E. coli ATCC 11775, E. faecalis ATCC 10541, K. pneumonia ATCC 13883, P. aeruginosa ATCC 27853, S. aureus ATCC 25922, and S. enterica ATCC 13076. All the bacterial strains were obtained from the American Type Culture Collection (Rockville, USA). The antibacterial assay was carried out as described in the literature [23]. The preparation of bacterial inocula was done by using 18 hold overnight bacterial cultures prepared in Nutrient Agar. A few colonies of bacteria were collected aseptically with a sterile loop and introduced into 10 mL of sterile 0.90 % saline solution. The concentration of the suspension was then standardized by adjusting the optical density to 0.10 at 600 nm, corresponding to bacterial cell suspension of 108 colony- forming units/mL (CFU/mL) [24]. This cell suspension was diluted 100 times to obtain 106 CFU/mL before use. The compounds were dissolved in DMSO and then added to bacteria suspension to obtain the final concentration of 5 % (v/v) DMSO or less. Serial twofold dilutions from 200 μg/mL of the compounds were performed in 96-well micro-titer plates. Each well contained 100 μL of sample of each concentration. Then each well was infunded 100 μL of the bacterial suspension. The final concentration range of the test compounds was 100–0.781 μg/mL, and the plates were incubated at 37 °C for 24 h. After incubation, the wells were examined for growth of microorganisms by measuring optical density (OD) value of the wells. Each experiment was repeated three times and Norfloxacin, bacteria suspension of 5 % (v/v) DMSO were used as a positive control and a blank control, respectively. By comparing to OD values, we can point out MIC values of these compounds among the selected concentration range.
References
K. Ponnusamy, C. Petchiammal, R. Mohankumar, W. Hopper, J. Ethnopharmacol. 132, 349–354 (2010)
A. Escalante, M. Gattuso, P. Perez, S. Zacchino, J. Nat. Prod. 71, 1720–1725 (2008)
R. Nair, T. Kalariya, C. Sumitra, Turk. J. Biol. 29, 41–47 (2005)
I. Weitzman, R.C. Summerbell, Clin Microbiol. Rev. 8, 240–295 (1995)
B.D. Ananth, U. Maheswara, C. Reddy, J. Pharm. Sci. Technol. 2, 119 (2010)
G.M. Cragg, D.J. Newman, K.M. Snader, J. Nat. Prod. 60, 52–60 (1997)
J.M. van der Nat, W.G. van der Sluis, K.T. de Silva, R.P.J. Labadie, Ethnopharmacol. 35, 1–24 (1991)
Q.G. Tan, X.D. Luo, Chem. Rev. 111, 7437–7522 (2011)
X.D. Luo, S.H. Wu, Y.B. Ma, D.G. Wu, Fitoterapia 71(6), 669–673 (2000)
X.D. Luo, Y.B. Ma, S.H. Wu, D.G. Wu, J. Nat. Prod. 62(7), 1022–1024 (1999)
K. Almas, Indian J. Dent. Res. 10(1), 23–26 (1999)
M. Jahan, M.K. Warsi, F. Khatoon, H. Hamid, Int. J. Res. Chem. Environ. 2(1), 33–37 (2012)
M. Bokel, R. Cramer, H. Gutzeit, S. Reeb, W. Kraus, Tetrahedron 46(3), 775–782 (1990)
W. Kraus, R. Crmer, Chem. Ber. 114, 2375–2381 (1981)
M. Takagia, Y. Tachia, J. Zhanga, T. Shinozakia, K. Ishiia, T. Kikuchia, M. Ukiyaa, N. Bannob, H. Tokudac, T. Akihisa, Chem. Biodivers. 11, 451–468 (2014)
R.E. Marker, H.M. Crooks, E.L. Wittbecker, J. Am. Chem. Soc. 63, 777–779 (1941)
S.B. Wu, Y.P. Ji, J.J. Zhu, Y. Zhao, G. Xia, Y.H. Hu, J.F. Hu, Steroids 74(9), 761–765 (2009)
H.Q. Duan, Y. Takaishia, H. Momota, Y. Ohmotob, T. Taki, Phytochemistry 59, 85–90 (2002)
K. Hisahiro, B. Masaki, O. Toru, Chem. Pharm. Bull. 55(1), 133–136 (2007)
X.L. Zhao, Z.M. Wang, X.J. Ma, W.G. Jing, A. Liu, China J. Chin. Mater. Med. 38(5), 703–708 (2013)
K.R. Markham, Tetrahedron 34(9), 1389–1397 (1978)
X.J. Qin, P.K. Lunga, Y.L. Zhao, J.L. Li, X.W. Yang, Y.P. Liu, X.D. Luo, Fitoterapia 92, 238–243 (2014)
S.M. Newton, C. Lau, S. Gurcha, G.S. Besra, C.W. Wright, J. Ethnopharmacol. 79, 57–67 (2002)
M.L. Tereshuck, M.V.Q. Riera, G.R. Castro, L.R. Abdala, J. Ethnopharmacol. 56, 227–232 (1997)
Acknowledgments
The authors are grateful to the Natural Science Foundation of China (81225024), the National Science and Technology Support Program of China (2013BAI11B02), for partial financial support, and to the analytical group of the Laboratory of Phytochemistry, Kunming Institute of Botany, for the spectral measurement.
Conflict of interest
The authors declare that there are no conflicts of interest.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
This article is published under license to BioMed Central Ltd. Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Liu, L., Zhao, YL., Cheng, GG. et al. Limonoid and Steroidal Saponin from Azadirachta indica. Nat. Prod. Bioprospect. 4, 335–340 (2014). https://doi.org/10.1007/s13659-014-0042-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-014-0042-2