
Abstract
Apis mellifera is present in Rodrigues, an island in the South-West Indian Ocean. The history of the established honeybee population is poorly known, and its biodiversity has never been studied. In this study, maternal origins of A. mellifera in Rodrigues have been assessed with the DraI test and sequencing of the mitochondrial COI-COII region. Nuclear genetic diversity was investigated with 18 microsatellite markers. A total of 524 colonies were sampled from 16 beekeepers in 20 sites. The Rodrigues population was characterized by the absence of the African lineage and presence of three European haplotypes from the C lineage. C1 haplotype was the most frequent (81.3 %), followed by C2 (18.0 %) (characteristics of A. m. ligustica and carnica, respectively) and a new haplotype, C1-Rod (0.7 %). No genetic structure was detected, and genetic diversity was comparable to continental populations. According to approximate Bayesian computation (ABC) analyses, bottleneck scenarios are most likely to have occurred.
Similar content being viewed by others


1 Introduction
During the past decade, honeybee populations have experienced declines and colony losses are continually reported in Europe and the USA (van Engelsdorp and Meixner 2010; Chauzat et al. 2013). The reduction of genetic diversity due to beekeeping practices is one of the several causes suggested (Oldroyd 2007) and has had an important impact on managed colonies (De la Rúa et al. 2013). Studies have demonstrated that reduced levels of intra-colonial genetic diversity affect colony fitness (Tarpy et al. 2013).
In its endemic area, Apis mellifera has evolved into at least 28 described subspecies (Ruttner 1988; Sheppard and Meixner 2003; Meixner et al. 2011, 2013) which have been grouped into four evolutionary branches using morphometric analysis (Ruttner 1988): the European North-Mediterranean (C) and the West-Mediterranean (M) lineages, the Oriental O lineage and the African A lineage. The breakdown of the different evolutionary lineages is supported by molecular analyses (Garnery et al. 1992; Whitfield et al. 2006; Wallberg et al. 2014). The European subspecies Apis mellifera ligustica and Apis mellifera carnica (C lineage) have been introduced worldwide because of their commercially desirable traits and have succeeded in adapting to new habitats (Chapman et al. 2008; Delaney et al. 2009).
In the South-West Indian Ocean, Apis mellifera unicolor (Latreille 1804) has been described as an endemic subspecies from Madagascar and belongs to the African lineage (Ruttner 1988). Honeybees are also established in the Mascarene archipelago (East of Madagascar) comprising La Réunion, Mauritius, and Rodrigues (Ruttner 1988). Rodrigues was discovered early in the sixteenth century, on the route to India (North-Coombes 1971). It is not known whether A. mellifera spp. were already present on the island before colonization. Controlled imports of honeybee queens are thought to have started in the 1930s, and around 1500 traditional hives were identified in the 1940s and 1950s (Bappoo and Ramanah 1989). In 1981, 11 European queens were reported to have been introduced to Rodrigues from the USA (Bappoo and Ramanah 1989). In 2002, the Rodrigues government restricted honeybee importation (Rodrigues apicultural services, unpublished data). In 2010, 2251 managed colonies from 173 beekeepers were identified (Belmin 2010). Since human colonization of the island, extensive habitat degradation has occurred with the total loss of the original forest (Thébaud et al. 2009).
In the Mascarene, mitochondrial diversity of honeybees had been investigated in La Réunion (n = 20) and Mauritius (n = 10) and AI was the sole sublineage detected (Franck et al. 2001). This study aims to describe the genetic diversity of the honeybee population in Rodrigues using molecular tools. First, the evolutionary lineages and subspecies were investigated using the mitochondrial COI-COII intergenic region. Then, the level of nuclear genetic diversity and its structure was investigated with microsatellite markers. Finally, we attempted to detect a bottleneck event, date it, and estimate effective population size before, during, and after the event.
2 Materials and methods
2.1 Sampling and DNA extraction
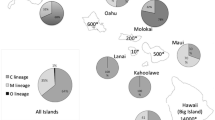
The study was conducted on the island of Rodrigues (18.3 km long and 8.5 km wide) (Figure 1). In February 2013, a total of 524 managed colonies belonging to 16 beekeepers were sampled from 20 different sites (Figure 1). One worker was collected from the entrance to each colony. Honeybees were preserved in ethanol (95 %) and kept at −20 °C until molecular analysis. Each worker was analyzed for both mitochondrial and microsatellite markers. Total DNA isolation was performed from the six legs of each individual following the method of Garnery et al. (1993) with slight modifications.

a Distribution of the mitochondrial sequences on the 20 sampled sites of Rodrigues. Number of individuals sequenced by site is indicated in the top of each diagram. b Details on beekeeper apiaries.
2.2 Mitochondrial DNA analysis
Mitochondrial analysis was carried out following Garnery et al. (1993). This consisted of PCR amplification of the intergenic COI-COII region using E2 and H2 primers, followed by a restriction of the amplified product with the DraI enzyme (Promega). PCR reaction was performed in a volume of 15 μL containing 2 mM MgCl2, 2 mM dNTPs, buffer 5× (Promega), 20 pmol of each primer, 30 units of GoTaq Flexi® Promega, 0.5 μL of DNA at 5 ng/μL. Samples underwent initial denaturation for 5 min at 92 °C, 35 cycles of 30 s at 92 °C, 45 s at 48 °C, 2 min at 62 °C, and a final elongation step of 7 min at 92 °C. DraI digestion was performed according to the manufacturer’s recommendations (Promega). Restriction fragments were separated in 5 % agarose MetaPhor gels.
The mitochondrial DNA (mtDNA) intergenic COI-COII region was sequenced in at least 30 % randomly selected individuals within each sample site, using the same E2-H2 primers. Individuals exhibiting rare restriction profiles were systematically sequenced. Sequence checking and alignment was done using Mega 5.1 software (Tamura et al. 2011) and then blasted on NCBI GenBank. All newly detected sequences were submitted to the GenBank database.
2.3 Microsatellite DNA analysis
A total of 18 polymorphic microsatellite loci (Estoup et al. 1994, 1995; Franck et al. 1998; Solignac et al. 2003) were scored A113, A24, AC306, AP55, AP81, A107, A29, A88, AP273, A28, AP289, B124, A35, A8, AP33, AP43, AP66, and A43 (Table S1). Primers were distributed into four multiplex PCR reactions (Table S1). PCR reactions were performed in 10-μL volumes containing 5 μL of Master Mix Type-it 2× Qiagen, 0.2 μL of each primer at 20 pmol/μL, and 1 μL of DNA at 5 ng/μL. All programs started with a denaturing cycle at 94° for 5 min, followed by 35 cycles of 30 s at 94 °C, 30 s at 52 °C, 45 s at 72 °C, and a final elongation at 72 °C for 20 min, except for Mix 1 where annealing was done at 55 °C. The samples were run through DNA sequencer ABI Prism 3130×L, and alleles were scored using Genemapper 4.0 (Applied Biosystems).
As a preliminary analysis, Micro-Checker 2.2.3 (Van Oosterhout et al. 2004) was used to identify genotyping errors. The frequency of null alleles per locus was estimated with FreeNa (Chapuis and Estoup 2007) based on 1000 bootstrap replicates. For each site with N ≥ 10 colonies, microsatellite diversity was estimated through mean number of alleles (N a ), observed (H obs ), and unbiased expected heterozygosity (H e ) (Nei 1978) with GENETIX 4.05 (Belkhir et al. 1996). Allelic richness (Ar) per site was calculated with FSTAT 2.9.3.2 (Goudet 2001) using the rarefaction method. Heterozygosity excess or deficit was estimated by calculating F IS according to Weir and Cockerham (1984). Deviations from the Hardy-Weinberg equilibrium (HWE) and linkage disequilibrium were tested using GENEPOP 3.4 (Raymond and Rousset 2004). False discovery rate (FDR) estimators (q-value) were calculated for probability values of repeated comparisons, using the R package QVALUE (Dabney et al. 2004). Pairwise F ST values were calculated for all combinations of the 18 sites (with N ≥ 10 colonies) and tested using permutations with FSTAT (Goudet 2001). Population structure and assignment of individuals to clusters were inferred using a Bayesian model-based clustering method with STRUCTURE 2.3.3 (Pritchard et al. 2000). Analyses were based on the admixture model with correlated allele frequencies with simulations of 105 burn-in steps and 106 iterations of MCMC algorithm for each run. Ten runs for each K (number of genetic clusters) value [1–20] were computed to estimate the most likely value of K. Optimal number of clusters was determined using Evanno’s estimator ∆K (Evanno et al. 2005) implemented in STRUCTURE HARVESTER (Earl and vonHoldt 2011). Principal components analysis (PCA) was used to visually identify and describe clusters of genetically related individuals with adegenet 1.3-9.2 package (Jombart 2008).
2.4 Testing for a bottleneck event using ABC
Approximate Bayesian computation (ABC) using DIYABC software version 2.0 was performed on microsatellite data to test for potential bottleneck events (Cornuet et al. 2008, 2010). This method ranks scenarios based on their approximate posterior probabilities. For each scenario, a large number of simulated data sets are produced by sampling parameter values into prior distributions (Table I). The occurrence of each scenario among the simulated data sets that are closest to the observed data gives an estimate of its posterior probability using a logistic regression procedure. Four demographic scenarios were compared here (Figure 2), two with a bottleneck and two without. In scenario 1, the Rodrigues population is assumed to have an effective number of individuals N 1 that has been constant over time, including at colonization. In scenario 2, an effective number of queens N b sampled from an ancestral population of effective size N anc , founded the Rodrigues population t generations ago. This bottleneck lasted d b generations. After that, the effective population size increased to the current population size N 1. It is reasonable to assume that the Rodrigues population was not founded from a single population source, but rather by several successive introductions from distinct origins. Such admixed origin may leave important signatures in the genetic structure of the population and should therefore be accounted for (Estoup and Guillemaud 2010). Scenarios 1 and 2 were complemented with two others accounting for the admixture at the origin of the Rodrigues population. In scenario 3, the current population resulted from the admixture of two ancestral and unsampled populations of sizes N 2 and N 3, respectively (same evolutionary lineage or not), at a time t a and with admixture rate r a . These two ancestral populations were assumed to have diverged from a common ancestor at date t 1. Scenario 4 is the same, except that during the admixture, the population suffered a bottleneck. The details on the method used for DIYABC estimations are given in Figure S1.

Schematic drawing of the four scenarios used for the ABC method.
3 Results
3.1 Mitochondrial DNA
PCR amplifications of the mtDNA intergenic COI-COII region were successful for 468 out of the 524 individuals sampled. All amplified products presented the same size ∼570 bp, which corresponds to the length of Q-type sequence, characteristic of the East European C lineage (Garnery et al. 1998). After PCR-RFLP analyses were performed, only one restriction fragment pattern was observed (fragment size ∼40/41, 47, 64, and 420 pb). Of the 278 individuals sequenced (Figure 1), three different sequences were detected. Two were previously described as the C1(a) haplotype belonging to populations of the A. m. ligustica subspecies (GenBank accession FJ478010.1 (Franck et al. 2001), JQ977699.1 (Muñoz et al. 2012), EF033655.1 (Collet et al. 2006)) and the C2(j) haplotype reported in A. m. carnica (JF723978.1). The third sequence exhibited 99 % similarity with C1 and presented a new polymorphic site. This new sequence was named C1-Rod (KF955993). C1 was the most frequent (n = 226, 81.3 %, KF955992) and was present in all sites; C2 (KF955994) was observed in 50 colonies from 11 sites (18.0 %), while C1-Rod was only detected in two colonies (0.7 %) of the same site S15 (Figure 1).
3.2 Microsatellite loci
A107 and AP81 (i) were difficult to score, (ii) presented high levels of null alleles (13.2 and 9.2 %, respectively), and (iii) had the highest levels of missing data (3.8 and 1.5 %, respectively). For these reasons, A107 and AP81 were removed from nuclear analysis. All individuals were successfully genotyped with at least ten loci amplified. Only nine loci combinations of 120 were significantly linked and involved different loci.
The number of detected alleles per locus ranged from three (AP273) to 15 (A029) for all individuals (n = 524) (Table S1). Over all the sites, the mean number of alleles (n = 16 loci) detected was 7.63 (±3.05). Genetic diversity between sites was comparable when sample size was corrected with an allelic richness ranging from 4.75 (±1.58) for S13 to 5.09 (±1.38) for S8. An asymptotic relationship was observed between the mean number of alleles and the cumulated number of sampled colonies (Figure S2). According to Pons and Chaouche (1995), the genetic diversity of the sample (representing 23.3 % of the colonies identified in 2010 (Belmin 2010)) was considered representative of the global genetic diversity of the Rodrigues honeybee population (Figure S2). Overall observed heterozygosity was similar to what was predicted under the HWE model (Table II). Multilocus values of F IS per site ranged from −0.075 (B1-b) to 0.051 (B6), and overall value was −0.001 (Table II). After FDR correction, none of these F IS values were significant and all sites were assumed to be under the HWE.
Genetic differentiation between sites (pairwise F ST) was low and ranged from −0.009 (sites S17 and S16) to 0.022 (sites S3 and S6). After Bonferroni corrections, only one pair of 153 pairwise comparisons was significant (F ST = 0.014; p value = 0.000260 < p value adjusted for multiple comparisons = 0.000263) for sites S15 and S12, respectively.
Analyses of Bayesian model-based clustering with STRUCTURE did not reveal evidence of population structure and differentiation, using the method of Evanno et al. (2005) (Figure S3). When K was >1, the probability of posterior assignment of each individual was equivalent for each cluster. Those results were supported by complementary PCA analyses which did not show any differentiation between groups of individuals.
3.3 Testing for a bottleneck event using ABC
Scenarios were ranked according to their posterior probabilities as follows: the best scenario was scenario 2 with a posterior probability of 0.558 (95 % confidence interval [0.543, 0.571]). Then, scenario 4 (p = 0.347 [0.333, 0.361]), followed by scenario 3 (p = 0.089 [0.080, 0.099]). The last was scenario 1 (p = 0.006 [0.000, 0.014]). Based on posterior probabilities, scenarios implicating a bottleneck (2 and 4) were more probable (a total posterior probability of 0.904) and supplanted the scenarios without a bottleneck (1 and 3, for a total posterior probability of 0.096). Full results of simulations to test for scenario confidence are given in Table S2. The probability that data sets simulated under scenarios without bottleneck (1 or 3) were assigned to the scenarios with bottleneck (2 or 4) amounted to 0.101 (type II error rate).
Parameter estimates, relative bias, and relative precision indices for scenario 2 are listed in Table I. Genetic parameters and ancestral population size N anc could not be estimated with precision (data not shown). Despite this, it is remarkable that noteworthy parameters could be estimated with satisfactory precision. The current population effective size N 1 was estimated at around 760 individuals (95 % confidence interval [333–1840]). The population experienced a bottleneck event 49 generations ago, ending 19 [7–95] generations ago and lasting approximately 30 [9–47] generations. Estimations suggested that around 60 [20–95] individuals colonized or were introduced to the island from a larger and ancestral population with an effective size of over 7463 [5470–9840].
4 Discussion
Mitochondrial analyses showed that all sampled colonies had only the Q sequence characteristic of the C lineage. This result is startling knowing that the honeybees of the surrounding islands are of African lineage (Ruttner 1988; Franck et al. 2001). Furthermore, Rodrigues mainly depends on Mauritius for commercial exchanges; honeybees could have easily colonized Rodrigues. European queens introduced in 1981 could have been more competitive and/or more favored by the beekeepers than the African lineage (if present).
The sequencing of the COI-COII intergenic indicated A. m. ligustica (C1, C1-Rod) and A. m. carnica (C2) descendants (Garnery et al. 1993, 1998). A. m. ligustica maternal lineages were four times more prevalent than A. m. carnica over all the sampling. Presence of European C lineage haplotypes is consistent with the reported introduction of queens bought from the US commercial stock (Bappoo and Ramanah 1989). In the USA, haplotypes C1 and C2 are characteristic of the most popular honeybee strains sold (Delaney et al. 2009). These commercially widespread subspecies have adapted well to the environmental conditions of this tropical island and persisted for over 32 years.
Furthermore, results of the ABC scenario choice support the assumption that the introduction of honeybees to the island was associated with a genetic bottleneck. Results from the bottleneck test are entirely credible in such insular ocean environments where colonization is limited by geographical barriers. According to the best scenario (2 with p= 0.558), this bottleneck event occurred approximately 49 generations ago (in an interval of 16 to 142 generations). One generation of A. mellifera is assumed to be approximately 2 years (Estoup et al. 1995; Excoffier et al. 2005). Present estimations are consistent with historical records of introductions of European honeybees in 1981. DIYABC results also suggest that the origin population had a large effective size (more than 7463 individuals). In the USA, the honeybee population was estimated to be 4.5 million colonies in 1980 (van Engelsdorp et al. 2008).
Nuclear genetic analyses indicated that the honeybee colonies on Rodrigues constituted one genetic population with homogenous diversity among sites. This observation was also demonstrated in other oceanic islands such as Menorca (701.8 km2) and Ibiza (572 km2) (De la Rúa et al. 2003). No genetic differentiation appeared along geographical distances indicating that gene flow is sufficient to homogenize nuclear genetic diversity. Rodrigues is a small island (104 km2) where apiary transhumance is not practiced by beekeepers. The distance between sedentary colonies might be too small to create genetic isolation (0.60 to 13.8 km). Exchange of queens between beekeepers, colony swarming, and male flight most likely ensure gene flow. Here, no nuclear genetic differences were found between colonies with the mtDNA sequence characteristics of A. m. ligustica and A. m. carnica whereas Muñoz et al. (2009) were able to detect differences between subspecies. This absence of genetic difference suggests that either hybrids were introduced or hybridization occurred after introduction. The study gives evidence of anthropogenic effects on the honeybee population in Rodrigues such as (i) the presence of exotic subspecies favored by beekeepers, (ii) hybridization phenomena between subspecies, and (iii) absence and/or non-utilization of the A lineage honeybees present in the area.
The Rodrigues honeybee population has levels of genetic diversity comparable to continental European populations of A. m. ligustica and carnica. For four comparable loci (A113, Ap55, B124, and Ap43), indigenous populations of A. m. carnica and/or ligustica in Croatia and Italy showed between 9.25 and 9.75 alleles per locus whereas 8.50 alleles were detected in the Rodrigues population (Muñoz et al. 2009). Another indication of the high levels of genetic diversity was heterozygosity (H obs = 0.644) which was equivalent to (i) indigenous Italian and Croatian populations (H obs = 0.638 and H obs = 0.674, respectively (Muñoz et al. 2009)) and (ii) US commercial populations (H obs = 0.53 in 2005 (Delaney et al. 2009)). Levels of heterozygosity of the Rodrigues honeybee population were higher than other insular systems in which A. m. ligustica was introduced. As an example, in the Azores, 35 % of colonies had haplotype C1 (cohabiting with 48 % of sublineage AIII and 17 % AII) and levels of observed heterozygosity were two times lower (H obs = 0.22 ± 0.03) than those in Rodrigues (De la Rúa et al. 2006). The same can be found in the Canary Islands, with only 31.2 % of observed heterozygotes in El Hierro (n = 17, 18 % of haplotype C1 and 82 % haplotype A15) and 41.3 % in Tenerife (n = 76, 35 % of C1 and three other African haplotypes) (De la Rúa et al. 2001). Unlike the rest of the world, the honeybee population in Rodrigues did not experience strong biological pressure caused by parasites and pathogens and this could explain these differences (vanEngelsdorp and Meixner 2010). A. mellifera in Rodrigues Island has a remarkable sanitary situation with no Varroa destructor (Belmin 2010), which was recently reported in Madagascar (Rasolofoarivao et al. 2013). To this day, the only known parasites and pathogens are the bee louse Braula pretoriensis and Nosema apis (Rehm 1988).
This study revealed a surprising lack of African lineage and exclusive presence of two subspecies: A. m. carnica and A. m. ligustica from the European C lineage. The honeybee population in Rodrigues did not show genetic structure but presented high levels of heterozygosity.
References
Bappoo, D.R., Ramanah, D. (1989) The state of beekeeping in Mauritius and other Mascarene Islands, p. 2. Proceedings Fouth International Conference on Apiculture in Tropical Climates, Cairo, Egypt
Belkhir, K., Borsa, P., Chikhi, L., Raufaste, N., Bonhomme, F. (1996) GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations. Laboratoire génome, populations, interactions. CNRS UMR 5000, 1996–2004
Belmin, R. (2010) Intérêt et faisabilité d’une Indication Géographique pour le miel de Rodrigues, p. 72. Université de Nancy, Nancy
Chapman, N.C., Lim, J., Oldroyd, B.P. (2008) Population Genetics of Commercial and Feral Honey Bees in Western Australia. J. Econ. Entomol. 101(2), 272–277
Chapuis, M.-P., Estoup, A. (2007) Microsatellite null alleles and estimation of population differentiation. Mol. Biol. Evol. 24(3), 621–631
Chauzat, M.P., Cauquil, L., Roy, L., Franco, S., Hendrikx, P., et al. (2013) Demographics of the European apicultural industry. PLoS One 8(11), e79018
Collet, T., Ferreira, K.M., Arias, M.C., Soares, A.E., Del Lama, M.A. (2006) Genetic structure of Africanized honeybee populations (Apis mellifera L.) from Brazil and Uruguay viewed through mitochondrial DNA COI-COII patterns. Heredity 97(5), 329–335
Cornuet, J.-M., Santos, F., Beaumont, M.A., Robert, C.P., Marin, J.-M., et al. (2008) Inferring population history with DIY ABC: a user-friendly approach to approximate Bayesian computation. Bioinformatics 24(23), 2713–2719
Cornuet, J.-M., Ravigné, V., Estoup, A. (2010) Inference on population history and model checking using DNA sequence and microsatellite data with the software DIYABC (v1. 0). BMC Bioinforma. 11(1), 401
Dabney, A., Storey, J.D., P.R.S. qvalue Tutorial, Install, R., Packages, I.B., et al. (2004) Q-value estimation for false discovery rate control. Medicine 344, 539–548
De la Rúa, P., Galián, J., Serrano, J., Moritz, R.F.A. (2001) Genetic structure and distinctness of Apis mellifera L. populations from the Canary Islands. Mol. Ecol. 10(7), 1733–1742
De la Rúa, P., Galián, J., Serrano, J., Moritz, R.F.A. (2003) Genetic structure of Balearic honeybee populations based on microsatellite polymorphism. Genet. Sel. Evol. 35(3), 339–350
De la Rúa, P., Galián, J., Pedersen, B.V., Serrano, J. (2006) Molecular characterization and population structure of Apis mellifera from Madeira and the Azores. Apidologie 37(6), 699–708
De la Rúa, P., Jaffé, R., Muñoz, I., Serrano, J., Moritz, R.F.A., et al. (2013) Conserving genetic diversity in the honeybee: comments on Harpur et al. (2012). Mol. Ecol. 22(12), 3208–3210
Delaney, D.A., Meixner, M.D., Schiff, N.M., Sheppard, W.S. (2009) Genetic Characterization of Commercial Honey Bee (Hymenoptera: Apidae) Populations in the United States by Using Mitochondrial and Microsatellite Markers. Ann. Entomol. Soc. Am. 102(4), 666–673
Earl, D.A., vonHoldt, B.M. (2011) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4(2), 359–361
Estoup, A., Guillemaud, T. (2010) Reconstructing routes of invasion using genetic data: why, how and so what? Mol. Ecol. 19(19), 4113–4130
Estoup, A., Solignac, M., Cornuet, J.-M. (1994) Precise assessment of the number of patrilines and of genetic relatedness in honeybee colonies. Proc. Roy. Soc. B Biol. Sci. 258(1351), 1–7
Estoup, A., Garnery, L., Solignac, M., Cornuet, J.-M. (1995) Microsatellite variation in honey bee (Apis mellifera L.) populations: hierarchical genetic structure and test of the infinite allele and stepwise mutation models. Genetics 140(2), 679–695
Evanno, G., Regnaut, S., Goudet, J. (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14(8), 2611–2620
Excoffier, L., Estoup, A., Cornuet, J.-M. (2005) Bayesian analysis of an admixture model with mutations and arbitrarily linked markers. Genetics 169(3), 1727–1738
Franck, P., Garnery, L., Solignac, M., Cornuet, J.-M. (1998) The origin of West European subspecies of honeybees (Apis mellifera): new insights from microsatellite and mitochondrial data. Evolution 52(4), 1119–1134
Franck, P., Garnery, L., Loiseau, A., Oldroyd, B.P., Hepburn, H.R., et al. (2001) Genetic diversity of the honeybee in Africa: microsatellite and mitochondrial data. Heredity 86(Pt 4), 420–430
Garnery, L., Cornuet, J.-M., Solignac, M. (1992) Evolutionary history of the honey bee Apis mellifera inferred from mitochondrial DNA analysis. Mol. Ecol. 1(3), 145–154
Garnery, L., Solignac, M., Celebrano, G., Cornuet, J.-M. (1993) A simple test using restricted PCR-Amplified mitochondrial DNA to study the genetic structure of Apis mellifera L. Experientia 49(11), 1016–1021
Garnery, L., Franck, P., Baudry, E., Vautrin, D., Cornuet, J.-M., et al. (1998) Genetic diversity of the west European honey bee (Apis mellifera mellifera and A. m. iberica). I. Mitochondrial DNA. Genet. Sel. Evol. 30, S31–S47
Goudet, J. (2001) FSTAT 2.9. 3.2, a program to estimate and test gene diversities and fixation indices
Jombart, T. (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24(11), 1403–1405
Latreille, P.A. (1804) Notice des espèces d’abeilles vivant en grande sociétée, ou d’abeilles proprement dites, et description d’espèces nouvelles. Ann. Mus. Natl. Hist. Nat. 5, 161–178
Meixner, M.D., Leta, M.A., Koeniger, N., Fuchs, S. (2011) The honey bees of Ethiopia represent a new subspecies of Apis mellifera—Apis mellifera simensis n. ssp. Apidologie 42(3), 425–437
Meixner, M.D., Pinto, M.A., Bouga, M., Kryger, P., Ivanova, E., et al. (2013) Standard methods for characterising subspecies and ecotypes of Apis mellifera. J. Apic. Res. 52(4), 1–27
Muñoz, I., Dall’Olio, R., Lodesani, M., De la Rúa, P. (2009) Population genetic structure of coastal Croatian honeybees (Apis mellifera carnica). Apidologie 40(6), 617–626
Muñoz, I., Stevanovic, J., Stanimirovic, Z., De la Rúa, P. (2012) Genetic variation of Apis mellifera from Serbia inferred from mitochondrial analysis. J. Apic. Sci. 56(1), 59–69
Nei, M. (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89(3), 583–590
North-Coombes, A. (1971) The island of Rodrigues. Mauritius, Published by the author, with the assistance of the Mauritius Advertising Bureau
Oldroyd, B.P. (2007) What’s killing American honey bees? PLoS Biol. 5(6), e168
Pons, O., Chaouche, K. (1995) Estimation, variance and optimal sampling of gene diversity II. Diploid locus. Theor. Appl. Genet. 91(1), 122–130
Pritchard, J.K., Stephens, M., Donnelly, P. (2000) Inference of population structure using multilocus genotype data. Genetics 155(2), 945–959
Rasolofoarivao, H., Clémencet, J., Ravaomanarivo, L.H.R., Razafindrazaka, D., Reynaud, B., et al. (2013) Spread and strain determination of Varroa destructor (Acari: Varroidae) in Madagascar since its first report in 2010. Exp. Appl. Acarol. 1–10
Raymond, M., Rousset, F. (2004) GENEPOP version 3.4. Population genetics software for exact tests and ecumenicism. Laboratoire de Genetique et Environment, Montpellier, France
Rehm, S.M. (1988) Report on the consultancy on bee diseases (Mauritius and Rodrigues), United nations development promgramme, pp. 77
Ruttner, F. (1988) Biogeography and taxonomy of honeybees. Springer-Verlag
Sheppard, W.S., Meixner, M.D. (2003) Apis mellifera pomonella, a new honey bee subspecies from Central Asia. Apidologie 34(4), 367–375
Solignac, M., Vautrin, D., Loiseau, A., Mougel, F., Baudry, E., et al. (2003) Five hundred and fifty microsatellite markers for the study of the honeybee (Apis mellifera L.) genome. Mol. Ecol. Notes 3(2), 307–311
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., et al. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28(10), 2731–2739
Tarpy, D.R., vanEngelsdorp, D., Pettis, J.S. (2013) Genetic diversity affects colony survivorship in commercial honey bee colonies. Naturwissenschaften 100(8), 723–728
Thébaud, C., Warren, B.H., Strasberg, D., Cheke, A. (2009) Mascarene islands. Biology 127, 1–216
Van Oosterhout, C., Hutchinson, W.F., Wills, D.P.M., Shipley, P. (2004) Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 4(3), 535–538
vanEngelsdorp, D., Meixner, M.D. (2010) A historical review of managed honey bee populations in Europe and the United States and the factors that may affect them. J. Invertebr. Pathol. 103, S80–S95
vanEngelsdorp, D., Hayes Jr., J., Underwood, R.M., Pettis, J. (2008) A Survey of Honey Bee Colony Losses in the U.S., Fall 2007 to Spring 2008. PLoS One 3(12), e4071
Wallberg, A., Han, F., Wellhagen, G., Dahle, B., Kawata, M., et al. (2014) A worldwide survey of genome sequence variation provides insight into the evolutionary history of the honeybee Apis mellifera. Nat. Genet. 46(10), 1081–1088
Weir, B.S., Cockerham, C.C. (1984) Estimating F-statistics for the analysis of population structure. Evolution 1358–1370
Whitfield, C.W., Behura, S.K., Berlocher, S.H., Clark, A.G., Johnston, J.S., et al. (2006) Thrice out of Africa: ancient and recent expansions of the honey bee, Apis mellifera. Science 314(5799), 642–645
Acknowledgments
We would like to thank J Félicité, G Volbert, and the Commission of Apiculture and Agriculture of Rodrigues for their welcoming and assistance in organizing the collection of samples. We are also grateful to the beekeepers: JJ Clain, JN Daya, L Felicité, D François, G François, J Herman-Casimir, JP Kan-John, JH Marianne, C & M Peermamode, MJ Ravanne, J Rikarl, and the feminine associations Ile Michel and Rodrigues-Farmer. We wish to thank C Simiand for his assistance in the laboratory. We would like to warmly thank V Ravigné for her precious help with DIYABC software; for the understanding of the methods, the analyses, and results; and for her corrections. We thank L Garnery for his advice on molecular methodology. We would like to thank the anonymous reviewers for improving the manuscript. Finally, we thank the network QualiREG and V Porphyre for helpful contacts. M A Techer is recipient of a PhD fellowship of the Sciences and Technology Doctoral School of the University of La Réunion. This study was partially funded by CIRAD, Région Réunion, and e-PRPV.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Manuscript editor: Marina Meixner
Caractérisation génétique de la population d’abeilles ( Apis mellifera ) de l’île Rodrigues, basée sur les microsatellites et l’ADN mitochondrial
analyse moléculaire / méthodes ABC / apiculture / tropiques / insularité
Genetische Charakterisierung der Honigbienenpopulation ( Apis mellifera ) auf der Insel Rodrigues, auf der Basis von Mikrosatelliten und mitochondrialer DNA
tropisch / insulär / molekulare Analysen / ABC-Methode / Bienenhaltung
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1010 kb)
Rights and permissions
About this article

Cite this article
Techer, M.A., Clémencet, J., Turpin, P. et al. Genetic characterization of the honeybee (Apis mellifera) population of Rodrigues Island, based on microsatellite and mitochondrial DNA. Apidologie 46, 445–454 (2015). https://doi.org/10.1007/s13592-014-0335-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13592-014-0335-9