
Abstract
Background
There are very few published data on the management of primary intrathoracic sarcoma, defined as sarcomas arising from the lung, pleura, and mediastinum, excluding the chest wall. Therefore, the aim of this study was to evaluate the outcome of patients with intrathoracic sarcoma treated at an academic referral center over a 21-year period.
Methods
A retrospective search was performed to identify patients with intrathoracic sarcomas treated with surgical resection from January 1990 to November 2011 at the University of Washington Medical Center. Local control and overall survival were analyzed in relation to the treatment received.
Results
Thirty-five patients were identified. Hazard ratios for local control, adjusted for tumor margin status, at 5 years were 0.74 (95 % CI [0.21, 2.58]) for the addition of chemotherapy (CT) to surgery, 0.57 (95 % CI [0.15, 2.23]) with the addition of (radiation therapy) RT, and 0.50 (95 % CI [0.06, 4.03]) with the addition of both CT and RT. At 7 years, the ratios for local control were 0.69 (95 % CI [0.20, 2.36]) for CT added to surgery, 0.58 (95 % CI [0.15, 2.27]) for RT, and 0.41 (95 % CI [0.05, 3.33]) with the addition of both CT and RT. Hazard ratios for overall survival, adjusted for sarcoma stage, at 5 years were 0.61 (95 % CI [0.16, 2.39]) for the addition of CT to surgery, 1.03 (95 % CI [0.26, 4.08]) for the addition of RT, and 0.54 (95 % CI [0.11, 2.69]) for the addition of both CT and RT. The 7-year hazard ratios for overall survival were 0.77 (95 % CI [0.23, 2.60]) for CT added to surgery, 0.99 (95 % CI [0.25, 3.84]) for the addition of RT, and 0.42 (95 % CI [0.09, 2.05]) for both CT and RT with surgery. At 10 years, hazard ratios for overall survival were 0.71 (95 % CI [0.21, 2.38]) for added CT, 0.81 (95 % CI [0.21, 3.08]) for added RT, and 0.33 (95 % CI [0.07, 1.65]) for the addition of both CT and RT to surgery.
Conclusions
Our series is the largest published study of intrathoracic sarcoma which focuses on the survival benefit of adding RT, chemotherapy or both to surgery in resectable intrathoracic sarcoma. Our data suggest a potential benefit in local control and survival from adjuvant therapy, with the greatest benefit likely to come from combined CT and RT, though none of the results achieved statistical significance. As intrathoracic sarcomas are rare and histologically heterogeneous, larger collaborative studies are necessary to determine treatment efficacy and elucidate which histologic subtypes are likely to benefit most from adjuvant therapy.
Similar content being viewed by others


Introduction
Sarcomas are malignancies that arise from cells of mesenchymal origin, and consequently can be found in bone, muscle, fat, cartilage and other connective tissue (http://cancer.gov/cancertopics/cancerlibrary/what-is-cancer). They are rare, accounting for less than 1 % of cancers in adults [1], and for approximately 15–20 % of tumors in children [2]. In addition these tumors are very heterogeneous with over 50 distinct histologic subtypes with a wide anatomical distribution [2]. Consequently due to this heterogeneity, studies of systemic therapy are very challenging. Historically they have been divided into soft tissue sarcomas, representing the majority of cases, and malignant bone sarcomas, which make up a little over 10 % [2].
Soft tissue sarcomas occur most commonly in the extremities, followed by the abdomen/pelvis and head and neck region [3]. In the USA, approximately 11,000 new cases are reported each year, and close to 4000 attributed deaths occur [4, 5]. They arise in all age groups, but are more common with age (51.7 % >60 years, 27.6 % 40–60 years, and 20.7 % 20–40 years), with a slight male predominance (1.1 male to 1.0 female) [6]. The etiology is usually unknown, though exposures to radiation, chemicals, and longstanding lymphedema have been suggested as risk factors [6]. Prognosis is most strongly correlated with histological grade [7, 8], and to a lesser extent with tumor size, positive surgical margins and histological subtype [9, 10].
Primary intrathoracic sarcomas are rare soft tissue sarcomas of the lung, pleura, and mediastinum. The most common histological subtypes are angiosarcoma, leiomyosarcoma, rhabdomyosarcoma, or a sarcomatoid variant of mesothelioma [11]. The rarity of these tumors is reflected in the paucity of literature specifically examining the efficacy of various treatment modalities to guide clinical therapeutic approaches. Frequently they are studied in combination with other soft tissue sarcomas, and often are not assessed as a distinct subset or not included at all [12–16]. They are commonly treated in a similar manner to soft tissue sarcomas of the extremities, for which more established guidelines exist [17].
Surgery is the primary treatment for localized intrathoracic sarcomas. The role of radiation and chemotherapy, however, is less established. Radiation therapy (RT) has been effective as an adjuvant to surgery for soft tissue sarcomas of the extremity [16, 18] and has been shown to reduce local recurrences with either pre- or post-surgery treatments [19]. The utility of chemotherapy (CT) is less clear, with no trial demonstrating a clear survival benefit in resectable soft tissue sarcoma [20–23]. Novel chemotherapeutic agents and combinations are currently being investigated in the metastatic setting, and may prove more efficacious [24]. Given the paucity of published data on the management of primary intrathoracic sarcoma, the purpose of this study was to assess the treatment and outcome of patients with intrathoracic sarcoma treated at the University of Washington, with particular attention to the effect of radiation and chemotherapy on local control and overall survival.
Research methods
Patient selection
IRB approval was obtained to review medical charts for patients diagnosed with thoracic tumors at the University of Washington Medical Center (UWMC) from January 1990 to November 2011. Patients identified with intrathoracic sarcomas via pathology and imaging, who received surgical resection with or without adjuvant CT and RT were included in the study. Patients who received initial incomplete resections elsewhere or who presented to our institution with locally recurrent disease were included, provided there was adequate information about their initial or primary tumor treatment, respectively. Over the time period analyzed, patients were treated by an experienced multi-disciplinary team of physicians. Informed consent for treatment was obtained from all patients included in this study. Neoadjuvant/adjuvant chemotherapy was administered based on individual patient presentation with particular attention to histological subtype, tumor grade, and tumor size.
Data collection
Resectable intrathoracic sarcoma was defined as a non-metastatic (stage I, II or III) primary sarcoma arising in the lung, pleura, or mediastinum. Each tumor was staged using the AJCC 7th edition staging criteria [25]. Patient demographic data (sex, age at diagnosis); tumor specific data (date of diagnosis, primary site, histology, size, grade, stage); margins status (positive <1 cm, negative >1 cm); treatment modalities received (neo-adjuvant radiation and/or chemotherapy or adjuvant radiation and/or chemotherapy); and outcomes (date of last follow-up, date of death) were obtained by retrospective chart analysis. The size of the tumor was defined as the maximum dimension (in centimeters) determined by CT at the initial diagnosis. All tumor pathology was examined centrally at the UWMC. Grade was classified according to the FNCLCC system as either low, intermediate or high-grade based on the number of mitoses, the amount of necrosis, and cellular differentiation [26].
Statistical analysis
Patients were followed from date of diagnosis to death of the patient, or end of the study period. Survival curves were plotted using the Kaplan-Meier method [27]. Hazard ratios for 5-, 7-, and 10-year local control and overall survival were obtained using Cox regression models [28], adjusting for tumor status (positive or negative) as a potential confounder in local control, and sarcoma stage (low or high) in overall survival. For the purposes of our analysis, “low stage” was defined as any stage I or II sarcoma, while “high stage” was defined as stage III. All analyses were conducted using R v.3.1.1 [29]. Local control was defined as the period from the date of first treatment to the date of first recurrence of disease at the original treatment site. Overall survival was defined as the interval between the first treatment and most recent follow-up or death.
Results
Patient population characteristics
Thirty-five adults with non-metastatic intrathoracic sarcomas treated at the University of Washington between January 1990 and November 2011 were identified. The patient population ranged in age from 19 to 75 years at the date of diagnosis (mean 47.4 years, median 49 years). There were 14 male patients (40 %) and 21 female patients (60 %). The site of tumor origin was the mediastinum in 15 cases (42.9 %), and pleura/lung in 20 cases (57.1 %). Tumor size was determined in 34 patients (97.1 %) and ranged in dimension from 2.7 cm to 33.5 cm (mean 12.3 cm). Common histological subtypes included synovial sarcoma (28.6 %), followed by leiomyosarcoma (14.3 %) and spindle cell sarcoma (11.4 %). There were 5 tumors classified as low grade (14.3 %), 12 as intermediate (34.3 %), and 18 tumors classified as high-grade (51.4 %). Five cases were categorized as stage 1B, 3 as stage IIA, 11 as stage IIb, and 16 as stage III, per the AJCC staging system. Patient characteristics, pathology, treatment and survival status are listed in Table 1.
Treatment characteristics
Twelve patients (34.3 %) received initial treatment elsewhere, prior to presenting at the UWMC. The previous treatments were excisional biopsy or surgical resection in 8 out of 12 patients (66.7 %), and surgical resection with preoperative chemotherapy in 4 patients (33.3 %). Of the 12 previously treated patients, 1 patient (8.3 %) presented with residual disease, 2 patients (16.7 %) presented with tumor recurrence, and 9 patients (75.0 %) had an alternative presentation (e.g., uncertain margin, negative margin). All 35 patients underwent surgical resection for their tumors, and postoperative margin status was recorded in all 35 cases. Of these patients, 2 (5.8 %) were left with gross disease, 20 patients (57.1 %) had a positive (<1 cm) margin and 13 (37.1 %) had a negative margin (≥1 cm). Fifteen patients (42.9 %) received surgery only, nine patients (25.7 %) received surgery and CT (either neo-adjuvant and/or adjuvant chemotherapy), eight patients (22.9 %) received surgery and RT (either neo-adjuvant and/or adjuvant radiation therapy), and 3 patients (8.6 %) received surgery and both CT and RT (either neo-adjuvant and/or adjuvant). The number of patients who received radiation was 11 (31.4 %) and the number of patients who received chemotherapy was 12 (34.3 %). Patients receiving chemotherapy primarily received ifosfamide and doxorubicin. One leiomyosarcoma patient also received gemcitabine and docetaxel and one of the synovial sarcoma patients received carboplatin and paclitaxel. Overall the treatment groups were similarly heterogeneous, with the exception of the three patients comprising the group that received all three treatment modalities (surgery + chemo + radiation) who were all stage III, high-grade synovial sarcoma cases.
Outcome
The mean patient follow-up time was 3.7 years. Disease recurrence was observed in 21 patients (61.8 %). Of these 21 patients, 12 (57.1 %) experienced local recurrence, 6 (28.6 %) experienced distant metastasis, and 3 (14.3 %) experienced both local recurrence and metastasis. The most common sites of metastases were the lung and bone (three patients each site) followed by the brain (twp patients). Patient status as of last follow-up revealed 9 patients (25.7 %) alive with no evidence of disease, 6 patients (17.1 %) alive with disease, 18 patients (51.4 %) had died of disease, and 2 patients (5.7 %) for whom the cause of death was unknown.
Local control and overall survival
Survival results comparing four treatment groups
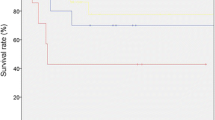
To compare the effects of different treatment patterns on overall and disease-free survival, the patient population was divided into four groups based on treatments received. Group 1 received surgery only, group 2 received surgery and CT, group 3 received surgery and RT, and group 4 received surgery and both CT and RT. The survival results are displayed as Kaplan-Meier curves in Fig. 1. Cox hazard ratio models comparing these treatment groups for 5-, 7-, and 10-year local control are detailed in Table 2. Hazard ratios for 5-, 7-, and 10-year overall survival are detailed in Table 3. Treatment with surgery alone is the reference group.

KM curves of overall survival and disease-free survival by 4-category treatment
Hazard ratios for local control at 5 years were as follows: 0.74 (95 % CI [0.21, 2.58]) for CT added to surgery, 0.57 (95 % CI [0.15, 2.23]) for RT, and 0.50 (95 % CI [0.06, 4.03]) with the addition of both CT and RT. At 7 years, the ratios for local control were 0.69 (95 % CI [0.20, 2.36]) for CT added to surgery, 0.58 (95 % CI [0.15, 2.27]) for RT, and 0.41 (95 % CI [0.05, 3.33]) with the addition of both CT and RT. 10-year local control hazard ratios were not calculated, given no events recorded after 7 years.
Hazard ratios for overall survival at 5 years were 0.61 (95 % CI [0.16, 2.39]) for the addition of CT to surgery, 1.03 (95 % CI [0.26, 4.08]) for the addition of RT, and 0.54 (95 % CI [0.11, 2.69]) for the addition of both CT and RT. The 7-year hazard ratios for overall survival were 0.77 (95 % CI [0.23, 2.60]) for CT added to surgery, 0.99 (95 % CI [0.25, 3.84]) for the addition of RT, and 0.42 (95 % CI [0.09, 2.05]) for both CT and RT with surgery. Hazard ratios for overall survival at 10 years were 0.71 (95 % CI [0.21, 2.38]) for CT added to surgery, 0.81 (95 % CI [0.21, 3.08]) for added RT, and 0.33 (95 % CI [0.07, 1.65]) for the addition of both CT and RT.
Survival results comparing radiation vs. no radiation
To examine the effect of RT on local control and overall survival, the patient population was divided into two treatment groups based on whether RT was administered (RT vs no RT). The survival results are displayed as Kaplan-Meier curves in Fig. 2. Cox hazard ratio models comparing these treatment groups for 5-, 7-, and 10-year local control and overall survival are detailed in Tables 4 and 5, respectively. The hazard ratios for local control at 5- and 7-years were 0.60 (95 % CI [0.18, 1.97]) and 0.59 (95 % CI [0.18, 1.91]), respectively, compared to surgery only. There were no reported events beyond 7 years. The hazard ratio for overall survival at 5 years was 0.90 (95 % CI [0.31, 2.68]), 0.71 (95 % CI [0.25, 2.04]) at 7 years, and 0.59 (95 % CI [0.20, 1.71]) at 10 years.

KM curves of overall and disease-free survival for addition of radiation therapy
Survival results comparing chemotherapy vs. no chemotherapy
To examine the effects of CT on local control and overall survival, the patient population was likewise divided into two groups (CT received vs. no CT). The survival results are displayed as Kaplan-Meier curves in Fig. 3. Cox hazard ratio models comparing these treatment groups for 5-, 7-, and 10-year local control and 5-, 7-, and 10-year overall survival are detailed in Tables 6 and 7. The hazard ratios for local control at 5 and 7 years were 0.78 (95 % CI [0.26, 2.35]) and 0.69 (95 % CI [0.24, 2.01]). For mortality at 5 years, the hazard ratio was 0.58 (95 % CI [0.19, 1.74]), 0.61 (95 % CI [0.22, 1.70]) at 7 years, and 0.56 (95 % CI [0.20, 1.56]) at 10 years.

KM curves of overall and disease-free survival for addition of chemotherapy
Discussion
Intrathoracic sarcomas are a rare subgroup of soft tissue sarcoma with poor prognosis and no established standard guidelines. Our study is the largest series of intrathoracic sarcoma treated at a single institution focused on treatment and outcomes of this rare set of neoplasms. This study highlights the variability in histology and individualization of patient treatment for this disease. Though surgery is the mainstay of therapy for localized sarcomas, recent studies indicate that the addition of (neo) adjuvant radiation and/or systemic therapy can potentially improve outcomes [3, 17, 30]. Similar studies examining thoracic wall (rather than primary pleural or pulmonary) sarcomas found that chemotherapy, radiation, and both in combination, each had a positive impact on local control and survival as adjunctive treatments [23, 31]. Our results, which are the first to address prognostic variables and treatment influence on local control and overall survival in intrathoracic sarcoma, are in agreement with these earlier findings for sarcomas presenting in other locations and underscore the prognostic power of high-grade disease in survival.
Notable limitations include lack of statistical power due to limited sample size and variability in treatment over the period of the study. However, our study is the first to compare these treatment modalities for intrathoracic sarcomas and attempt quantification of their effect on local control and survival. Despite the limitations, this study will serve as a benchmark for further larger studies evaluating novel approaches in the management of intrathoracic sarcoma.
Due to the numerous histological subtypes and manifestations of intrathoracic sarcomas, a multi-disciplinary approach to their management is essential, as coordination between surgery, radiation, medical oncology, pathology and radiology will facilitate clearer classification and management strategies for these rare malignancies [17]. In order to improve the outcome of patients with rare diseases, it is important that treatment is undertaken at specialist centers [32]. Furthermore, international collaboration is required to assess the utility of novel therapies in such malignancies as well as identifying the underlying molecular drivers of these heterogeneous diseases.
References
Howlader N, Noone AM, Krapcho M, Neyman N, Aminou R, Altekruse SF, Kosary CL, Ruhl J, Tatalovich Z, Cho H, Mariotto A, Eisner MP, Lewis DR, Chen HS, Feuer EJ, Cronin KA (eds). SEER Cancer statistics review, 1975–2009 (vintage 2009 populations), National Cancer Institute. Bethesda, MD, http://seer.cancer.gov/csr/1975_2009_pops09/, based on November 2011 SEER data submission, posted to the SEER web site, April 2012
Burningham Z, Hashibe M, Spector L, Schiffman JD (2012) The epidemiology of sarcoma. Clin Sarcoma Res 2:14. doi:10.1186/2045-3329-2-14
Alkis N, Muallaoğlu S, Koçer M, Arslan Y, Durnalı AG, Tokluoğlu S, Çelenkoğlu G, Paksoy F, Coskun U (2011) Primary adult soft tissue sarcomas: analysis of 294 patients. Med Oncol 28(1):391–6
American Cancer Society (2012) Cancer facts and figures 2012. Atlanta, Ga: American Cancer Society. Available online. Last accessed October 24
Siegel R, Naishadham D, Jemal A (2012) Cancer statistics 2012. CA Cancer J Clin 62(1):10–29
Pisters P, Weiss M, Maki R (2011) Soft-tissue sarcomas in cancer management: 14th edition. October 14, 2011 http://www.cancernetwork.com/cancer-management/soft-tissue-sarcomas/article/10165/1802713
Coindre JM, Terrier P, Guillou L, Le Doussal V, Collin F, Ranche’re D (2001) Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1,240 patients from the French federation of cancer centers sarcoma group. Cancer 91:1914–24
Coindre JM, Terrier P, Bui NB, Bonicho F, Collin F, Doussal VL et al (1996) Prognostic factors in adult patients with locally controlled soft tissue sarcoma. A study of 546 patients from the French federation of cancer centers sarcoma group. JCO 14:869–77
Gutierrez JC, Perez EA, Franceschi D, Moffat FL Jr, Livingstone AS, Koniaris LG (2007) Outcomes for soft-tissue sarcoma in 8249 cases from a large state cancer registry. J Surg Res 141(1):105–14
Singer S, Corson JM, Demetri GD et al (1995) Prognostic factors predictive of survival for truncal and retroperitoneal soft-tissue sarcoma. Ann Surg 221(2):185–95
Gladish GW, Sabloff BM, Munden RF, Truong MT, Erasmus JJ, Chasen MH (2002) Primary thoracic sarcomas. Radiographics 22(3):621–37
Cakir S, Dincbas FO, Uzel O et al (1995) Multivariate analysis of prognostic factors in 75 patients with soft tissue sarcoma. Radiother Oncol 37(1):10–16
Coindre JM, Terrier P, Bui NB et al (1996) Prognostic factors in adult patients with locally controlled soft tissue sarcoma. A study of 546 patients from the French federation of cancer centers sarcoma group. J Clin Oncol 14(3):869–77
Lehnert T, Schwarzbach M, Willeke F et al (2000) Intraoperative radiotherapy for primary and locally recurrent soft tissue sarcoma: morbidity and long-term prognosis. Eur J Surg Oncol 26(Suppl A):S21–4
Peiper M, Zurakowski D, Zornig C (1995) Local recurrence of soft tissue sarcoma of the extremities and trunk. Langenbecks Arch Chir 380(6):333–9
Pisters PW, Harrison LB, Leung DH, Woodruff JM, Casper ES, Brennan MF (1996) Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. J Clin Oncol 14(3):859–68
Levra MG, Novello S, Scagliotti GV, Papotti M, Le Cesne A (2012) Primary pleuropulmonary sarcoma: a rare disease entity. Clin Lung Cancer 13(6):399–407. doi:10.1016/j.cllc.2012.05.003
Yang J, Chang AE et al (1998) Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. JCO 16(1):197–203
O’Sullivan B, Davis AM (2002) Preoperative versus postoperative radiotherapy in soft tissue sarcoma of the limb; a randomized trial. Lancet 359:2235–2241
Woll PJ et al (2012) Adjuvant chemotherapy with doxorubicin, ifosfamide, and lenograstim for resected soft-tissue sarcoma (EORTC 62931): a multicentre randomized controlled trial. Lancet Oncol 13(10):1045–54
(1997) Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Sarcoma meta-analysis collaboration. Lancet; 350(9092):1647–54
Pervaiz N et al (2008) A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 113(3):573–81
Gronchi A et al (2013) Quality of surgery and neoadjuvant combined therapy in the ISG-GEIS trial on soft tissue sarcomas of limbs and trunk wall. Ann Oncol Off J Eur Soc Med Oncol ESMO 24:817–823
Constantinidou A (2013) The evolution of systemic therapy in sarcoma. Expert Rev Anticancer Ther 13(2):211–23
Edge SB, Byrd DR (2010) AJCC cancer staging manual. 7th ed. Springer Verlag
Costa J, Wesley RA, Glatstein EJ, Rosenberg SA (1984) The grading of soft tissue sarcomas. Results of a clinicohistopathologic correlation in a series of 163 cases. Cancer 53:530–41
Kaplan EL, Meier P (1958) Nonparametric estimation from incomplete observations. J Am Stat Assoc 53:457–481
Cox DR (1972) Regression models and life tables (with discussion). J R Stat Soc Ser B 34:187–220
R Core Team (2014) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL http://www.R-project.org/
Okby NT, Travis WD (2000) Liposarcoma of the pleural cavity: clinical and pathologic features of 4 cases with a review of the literature. Arch Pathol Lab Med 124:699–703
Burt A, Berriochoa J, Korpak A, Rodler E, Jones R, Loggers E, Weisstein J, Patel S (2013) Treatment of chest wall sarcomas: a single institution experience over twenty years. Am J Clin Oncol. Levra
National Comprehensive Cancer Network. Soft Tissue Sarcoma (version 2.2014). http://www.nccn.org/professionals/physician_gls/pdf/sarcoma.pdf. Accessed July 15, 2014
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Statement of ethical standards
This article does not contain any studies with human or animal subjects performed by any of the authors.
Funding sources
No funding support is associated with this study.
Financial disclosures
None.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rainey, D., Berriochoa, J., Korpak, A. et al. Treatment of resectable intrathoracic sarcomas: a single institution experience over twenty years. J Radiat Oncol 5, 169–177 (2016). https://doi.org/10.1007/s13566-015-0236-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13566-015-0236-1