
Abstract
The interactions between fibroblast growth factors (FGFs) and their receptors (FGFRs) are facilitated by heparan sulfate (HS) and heparin (Hp), highly sulfated biological polyelectrolytes. The molecular basis of FGF interactions with these polyelectrolytes is highly complex due to the structural heterogeneity of HS/Hp, and many details still remain elusive, especially the significance of charge density and minimal chain length of HS/Hp in growth factor recognition and multimerization. In this work, we use electrospray ionization mass spectrometry (ESI MS) to investigate the association of relatively homogeneous oligoheparins (octamer, dp8, and decamer, dp10) with acidic fibroblast growth factor (FGF-1). This growth factor forms 1:1, 2:1, and 3:1 protein/heparinoid complexes with both dp8 and dp10, and the fraction of bound protein is highly dependent on protein/heparinoid molar ratio. Multimeric complexes are preferentially formed on the highly sulfated Hp oligomers. Although a variety of oligomers appear to be binding-competent, there is a strong correlation between the affinity and the overall level of sulfation (the highest charge density polyanions binding FGF most strongly via multivalent interactions). These results show that the interactions between FGF-1 and Hp oligomers are primarily directed by electrostatics, and also demonstrate the power of ESI MS as a tool to study multiple binding equilibria between proteins and structurally heterogeneous polyanions.

ᅟ
Similar content being viewed by others
Introduction
Glycosaminoglycans (GAGs), in particular heparan sulfate (HS), and heparin (Hp), are engaged in a wide spectrum of physiological processes, including embryogenesis, immune response, cell proliferation, differentiation, and angiogenesis. All of these are related to the ability of these heparinoids to potentiate the activity of numerous signaling proteins (e.g., by mediating the interactions of growth factors and their cell surface receptors, or by sequestering and releasing signaling proteins in the extracellular matrix. The main challenge in characterizing GAG-protein binding arises from the immensely polydisperse GAG structure due to (1) wide distribution of chain lengths, and (2) apparently stochastic sulfation patterns. Although this polydispersity allows GAGs to interact with a variety of proteins, the details of the relationship between sulfation and protein affinity remain largely elusive. The fact that GAGs are the most highly charged macromolecules in animals suggests that electrostatic forces play significant roles in modulating their protein affinity, but such long-range interactions are typically relegated to supportive roles in protein recognition. Deciphering the structure–property relations of the heparinoids, which would remove a major barrier to the development of potential biomedical applications of these GAGs beyond their well-established role as anticoagulants, is thus coupled to the need to understand the role of sulfation patterns in binding their diverse physiological partners [1–4].
In the GAG family, HS is composed of repeating disaccharide units of N-acetylated (GlcNAc)/N-sulfated glucosamine (GlcNS) and glucuronic acid (GlcA) or iduronic acid (IdoA) [5]. The biosynthesis is not controlled by a genetic template and the polymer chain undergoes a series of enzymatic modifications (de-acetylation, and addition of sulfates at various positions on the polysaccharide backbone by N-sulfotransferases and O-sulfotransferases) and epimerization (transformation of glucosamine unit to L-iduronic acid). These modifications do not occur uniformly across the entire chain during biosynthesis of HS and Hp; for example, the structure of HS exhibits domains of high sulfation, rich in IdoA and O-sulfation (NS), non-sulfated but N-acetylated (NA) domains, and alternating NS/NA domains [6]. Hp is structurally similar to the extended NS domains of HS comprising regions of N- and O-sulfation, which are thought to be involved in protein binding events. This justifies the common use of Hp as a proxy for HS in studies of GAG–protein interactions, and their similarity prompted the use of the term “heparinoid” [7].
Growth factors (GFs) are signaling proteins that bind to their transmembrane receptors (GFRs) to initiate cell proliferation, differentiation, and angiogenesis [8]. The interaction between GF and GFR is promoted by cell surface HS, which reduces dimensionality of the protein/receptor encounter from three dimensions (for the free protein in solution) to one (for the GAG-bound state), which facilitates the docking process [9]. HS acts as a low affinity but abundant receptor for GFs and increases the possibility of finding less abundant high affinity receptor on the cell surface [10]; in vivo studies have provided strong evidence for the role of HS in effective FGF signaling [11–13].
Previous efforts to characterize HS and Hp interactions with growth factors have focused on the contribution of specific sulfate groups, such as 6-O-sulfates for FGF-10, 2-O sulfates for FGF-2, or both for FGF-4 and FGF-7 [14, 15]. The basic premise of this approach is that pair-matching of basic residues on the protein and sulfates on the heparin oligomers drives the interaction. However, in these studies, the importance of a more general charge complementarity between the protein and the polyelectrolyte-like HS/Hp had not been considered [16, 17], possibly overlooking the role of more long-range multivalent electrostatics in the dynamic recognition between the protein and the highly heterogeneous and flexible polyanionic chains.
All heparin-binding proteins appear to have well-defined positive domains within globally negative molecules, whereas GAGs usually have non-uniform structure [18]. There is growing evidence that strong binding occurs between globally negative proteins and polyanions when polyanion charge distributions are arranged in a way that minimizes long-range repulsion while optimizing short-range attractions with locally positive protein domains [19, 22]. This can result in a form of selectivity that does not arise from specific short-range interactions such as hydrogen bonding or salt bridge formation [20–22]. Supporting this perspective, several groups suggested nonspecific contributions to GAG interactions with growth factors. Catlow et al. showed that the selectivity of hepatocyte growth factor binding to HS is strongly influenced by HS sulfate density [23]. Krueger et al. indicated that various FGFs share the same binding domain on the HS, where binding affinity is correlated with the extent of sulfation [24]. Jastrebova et al. extended the studies to FGF-2 and its receptors, and revealed that highly sulfated chains induced ternary complex formation [25] and FGF-2 cellular signaling [26]. These findings point to the need for a new concept of GAG-protein specificity that considers charge complementarity between the protein and the related polyanion [27] beyond well-defined ion pairs (such as salt bridges in protein/protein and protein/nucleic acid interactions).
Although it is clear that the sulfation patterns are important determinants of polysaccharide-induced protein assembly, most of the studies of protein/GAG interactions cannot make a distinction among various polyanionic structures present in highly heterogeneous Hp or HS samples and, as a result, probe the behavior averaged across the entire ensemble of GAG oligomers. An earlier attempt to circumvent this problem by taking the advantage of the ability of mass spectrometry (MS) to make a distinction among various species based on differences in their masses failed to produce meaningful results for FGF/GAG association [28]. The use of matrix-assisted laser desorption ionization (MALDI) MS in that study prevented the observation of specific associations (even though there are several examples of successful use of MALDI MS to detect noncovalent biopolymer complexes [29], such studies typically require careful selection of experimental conditions to ensure that biopolymers are co-crystallized under non-denaturing conditions). Furthermore, MALDI is typically used with time-of-flight (TOF) mass analyzers, which provide high resolution in the low m/z region, but the resolution decreases dramatically at high m/z (typical for work with noncovalent complexes). This problem is further compounded by massive adduct formation and low detection efficiency for high-mass ions, unless the instrument is extensively modified [30]. Native electrospray ionization (ESI) mass spectrometry offers an attractive alternative vis-à-vis characterization of relatively short heparinoid/protein complexes, as it allows both the binding stoichiometry [31–33] and the overall degree of sulfation of the heparinoid component [34, 35] to be readily deduced from the mass of the complex. Furthermore, combination of native ESI MS with methods of ion manipulation in the gas phase (such as limited charge reduction [36]) enables meaningful analysis of protein interactions with much more heterogeneous GAGs, such as intact unfractionated heparin [37]. In this work we use native ESI MS to probe the ability of short heparinoids to induce multimerization of FGF-1 and examine the effects of the polyanion concentration in solution, its chain length, and the total extent of sulfation.
Experimental
Materials
The H93G mutant of the acidic fibroblast growth factor (FGF-1) was kindly provided by Professor Robert Linhardt (RPI, Troy, NY, USA). The mutation is introduced to increase stability [38] by shifting the protein pI to 7.8. Heparin oligomers (octa- and deca-saccharides, dp8 and dp10), prepared by partial heparin lysis followed by high resolution gel filtration, were generously donated by Dr. John Gallagher (Iduron, Manchester, UK). All other chemicals and solvents used in this work were of analytical grade or higher.
Methods
All ESI MS measurements were carried out with a QStar-XL (AB Sciex, Toronto, Canada) hybrid quadrupole-time-of-flight mass spectrometer equipped with a nano-ESI source. Both closed and pre-opened (1–2 μm id) glass nanospray capillaries (New Objective, Woburn, MA, USA) were used in this work. The FGF-1 solution concentration was kept constant throughout all measurements (0.08 g/L, or ca. 5 μM in 100 mM NH4CH3CO2 at pH 6.8), whereas the dp8/dp10 concentration was varied from 1 to 25 μM. The FGF-1 solution concentration was verified by measuring UV-VIS absorbance (using molar absorptivity of 17545 M−1 cm−1). Both dp8 and dp10 were diluted to the required concentration from 2 mg/mL stock solutions. To ensure stability of the protein/heparinoid complexes in the gas phase, all mass spectra were acquired using the following settings of ion optics elements in the ESI interface: DP, 100; FP, 265; and DP2, 15. The mass distributions of the protein-bound heparinoid molecules I(M dpX) were recalculated from the raw data I(m/z) using the following formula:
where z is the ionic charge, n is the number of protein molecules in the complex, and M protein is the mass of the neutral protein molecule (15892.9 Da).
Electrostatic potentials around FGF-1 (PDB id: 1K5U) were calculated at pH 7.0 and I = 100 mM using DelPhi V. 4r1.1 [39, 40], which applies nonlinear Poisson-Boltzmann equation to generate the potential surface of the protein. Fractional charges of amino acids on the protein surface were determined by fitting experimental pH titration curves, using the spherical-smeared charge model of Tanford and Kirkwood [41].
Results and Discussion
Factors Controlling Heparinoid-Assisted FGF-1 Multimerization
The ESI mass spectrum of FGF-1 acquired in the absence of oligoheparins showed no evidence of dimers or higher-order multimer formation (Figure 1). This is consistent with the results of SEC analysis of this sample showing a single symmetrical peak for monomer with no early eluting species that would be indicative of the presence of dimeric and multimeric forms of the protein (data not shown). However, formation of the protein oligomers was clearly visible when either dp8 or dp10 was present in solution with the protein at [FGF]/oligoheparin molar ratio r = 2 or higher. Binding of up to three FGF-1 molecules to a single heparin chain was observed (Figure 2), and FGF multimerization is clearly favored in solutions with higher protein/heparin molar ratio. Polyanion chain length promotes multimerization (e.g., compare the relative abundance of FGF3·dp8 and FGF3·dp10 ionic species in Figure 2). Formation of the 1:1 FGF/dp8 complex is strongly favored at low protein/heparin ratio (r = 0.2), whereas the 2:1 complex becomes prominent at r = 0.5 and is the most abundant species at higher values of r. This behavior is mirrored by dp10, which forms only 1:1 complexes with the protein at low r. As r increases, heparin-bridged dimers and trimers of FGF also appear in the mass spectra, the 2:1 complex being the most abundant ionic species at r = 2. It is important to note that this behavior (the increase of the extent of protein association upon decreasing the oligoheparin concentration while keeping the protein concentration constant) provides very strong evidence that the observed protein/oligoheparin assemblies originate from solution, rather than represent gas-phase artifacts (similar to those observed in MALDI MS analysis of FGF/oligoheparin interactions [28]). Indeed, should these complexes be formed in the ESI interface as a result of nonspecific interactions, one would expect to see a dramatic increase in complex ion formation upon a 10-fold increase of the oligoheparin concentration, exactly opposite to the trend shown in Figure 2.

ESI mass spectrum of a 5 μM solution of FGF-1 in 100 mM NH4CH3CO2 at pH 6.8

ESI mass spectra of FGF-1 incubated with dp8 (left panel) and dp10 (right panel) it 100 mM NH4CH3CO2 at pH 6.8. The numbers in boxes indicate charge states of free FGF-1 (black), and its complexes with heparinoids: 1:1 (red), 2:1 (blue), and 3:1 (green). The r values shown in each row indicate the protein/heparinoid molar ratio
Another intriguing feature of the ionic signals of FGF/dp8 and FGF/dp10 complexes is a noticeable variation in the widths of the ionic peaks reflecting structural heterogeneity of the heparin component of these complexes. The 1:1 complexes appear to display broader mass distributions, even after correction is made for the lower number of charges carried by these ions (higher number of charges compresses the mass distributions on the m/z scale). This could indicate that not all heparin chains are capable of binding multiple FGF-1, whereas many chains can provide binding sites for a single protein molecule, pointing towards the promiscuous nature of these interactions [42] (vide infra).
The 3:1 complex (already visible in the mass spectrum acquired at r = 2) is even more abundant at r = 4 (Figure 3), where the signal of the 1:1 complexes is nearly absent from the mass spectrum, and the ionic intensity is distributed almost uniformly among the FGF2·dp10 and FGF3·dp10 species. The inability of the abundant free FGF molecules in solution to drive the complexation process to complete saturation (i.e., convert all FGF2·dp10 complexes to the FGF3·dp10 form) provides additional evidence that only a subset of the entire pool of dp10 molecules have sulfation patterns that allow three protein molecules to be accommodated on a single oligoheparin chain.

ESI mass spectrum of dp10 incubated in a 5 μM solution of FGF-1/100 mM NH4CH3CO2 at pH 6.8 (protein/heparinoid molar ratio r = 4)
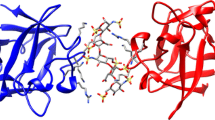
The observation of the 3:1 FGF/dp10 complexes raises an intriguing question: How can the relatively short oligomer chain accommodate three FGF molecules? Dynamic and static light scattering studies showed that 14–15 FGF-1 molecules could bind to a single intact Hp chain, averaging four monosaccharide units per protein molecule [43], suggesting that accumulation of up to three protein molecules per single dp10 chain might be problematic. To visualize these possibilities, the FGF-1 surface charge distribution in solution at pH 7.0 and ionic strength I = 100 mM was generated using DelPhi (Figure 4). The resultant electrostatic potential contours at 0.5 kT/e indicate a dominant positive patch at pH 7.0, which appears to be suitable for binding a polyanionic chain. Even though the exact location of the protein-bound chain cannot be determined, it can be assumed that the highly negative heparin would avoid the negatively charged protein domains. Furthermore, it seems unlikely that this association conforms to the classical “lock-and-key” interaction model. Instead, the oligoheparin is likely to be kept within the immediate vicinity of the positive protein domain, which generates a strong attraction basin for polyanions without forcing them to a specific highly defined (and, therefore, entropically costly) conformation. This dynamic behavior notwithstanding, if the FGF-binding site size is assumed to consist of four contiguous monosaccharide units (~2 nm [43]), binding of three FGF-1 molecules (hydrodynamic radius 2 nm) to a single dp10 chain (contour length 5 nm) could not realistically be accomplished in a cis configuration. However, it is possible that the semiflexible heparin chain [44, 45] can accommodate FGF-1 molecules in different directions, leading to formation of a trans complex (Figure 4). Importantly, multivalent protein binding to the heparinoid chain is likely to be governed not only by the electrostatic attraction between the polyanion and protein but also by repulsion among the protein molecules (the latter would dictate the “allowed” proximity between the two protein molecules located on the “same side” of the chain).

Delphi-generated electrostatic potential contours (–0.5 kT/e, red and +0.5 kT/e, blue) generated around FGF-1 (PDB id: 1K5U) at pH 7.0 and ionic strength I = 100 mM (top). A representative dp10 molecule (a deca-saccharide segment of 3IRJ) is shown for comparison to provide the physical dimensions of the polyanionic chain relative to the protein dimensions. Diagrams at the bottom show possible arrangements of dp10 (shown schematically as a ruler) and FGF-1 in multi-valent complexes
Binding Preferences of Heparin Oligomers: Correlation Between Charge Density and Affinity
The two heparinoids examined in this work, dp8 and dp10, have fixed lengths, but nonetheless exhibit structural polydispersity with respect to both total levels of sulfation and the distribution of the sulfate groups across the polysaccharide backbone (“sulfation patterns”). Observation of protein/heparinoid complexes of different stoichiometries complexes indicates the existence of multiple biding sites within each chain with a range of affinities for FGF-1. In order to identify the binding preferences of dp8 and dp10 species with different levels of sulfation, the molecular weight distributions of FGF-bound heparinoids were determined based on the shapes of the most abundant ionic peaks representing various protein/heparinoid complexes in ESI mass spectra shown in Figures 2 and 3. This was done by converting the mass-to-charge ratios to masses of the neutral complexes, followed by subtraction of the mass of the protein component using Equation 1. The results of these calculations show the mass distributions of dp8 (Figure 5) and dp10 (Figure 6) species participating in formation of the FGF/heparinoid complexes of different stoichiometries and at different mixing ratios. Since all analyses were performed in the positive ion mode, each ionic species is expected to incorporate 4–5 cations, such as Na+ or NH4 + in addition to H+, to neutralize the excessive negative charge on the heparinoid chain [35]. Given the uncertainty in the total number of charge-neutralizing cations attached to the polyanionic chain, and the mass difference between Na+ or NH4 +, the mass of a specific heparinoid molecule is not a specific number, but rather a range (shown in Figures 5 and 6 as a shaded a box for each species), even though both the length and the total number of sulfate groups are fixed. Although these ranges partially overlap for heparinoids whose extents of sulfation are similar, a clear distinction can be made between the species that differ by at least two sulfate groups (the notations in Figures 5 and 6 are based on the Roepstorff-Henriksen nomenclature, which identifies heparinoid molecules with three numbers, the number of saccharide units in the chain, the total number of sulfate groups, and the number of acetyl groups [46]). Overall, the extent of sulfation for the binding-competent dp8 species ranges from 8 to 12 (Figure 5), whereas the total sulfation range for binding-competent dp10 species ranges from 10 to 15 (Figure 6).

Mass distributions of the FGF-bound dp8 molecules at two different protein/heparinoid molar ratios. Mass distributions of the dp8 chains involved in formation of 1:1 complexes (red traces) are calculated based on the ionic signal of FGF·dp8+7 species, and those for 2:1 complexes (blue traces) are calculated based on the ionic signal of FGF2·dp8+12 species. The colored boxes indicate the mass ranges corresponding to the dp8 species with varying levels of sulfation

Mass distributions of the FGF-bound dp10 molecules at several different protein/heparinoid molar ratios. Mass distributions of the dp10 chains involved in the formation of 1:1 complexes (red traces) are calculated based on the ionic signal of FGF·dp10+7 species, those for 2:1 (blue traces), and 3:1 (green trace) complexes are calculated based on the ionic signals of FGF2·dp10+12 and FGF3·dp10+14 species, respectively. The colored boxes indicate the mass ranges corresponding to the dp10 species with varying levels of sulfation
The mass distributions of FGF-bound dp8 molecules (Figure 5) show a clear difference among the polyanionic chains accommodating only a single protein molecule (blue trace) and those bridging two proteins (pink traces). Not surprisingly, only the chains with a relatively high extent of sulfation participate in the formation of the 2:1 FGF/dp8 complexes: at least 10 sulfate groups are required for dp8 to bridge two FGF molecules, whereas the chains incorporating 11 or 12 sulfate groups give rise to the most abundant dp8-bridged FGF dimers (Figure 5). In contrast, 1:1 complexes readily form even when the total number of sulfate groups is eight. Comparison of the mass distributions acquired at different FGF/dp8 ratios provides unequivocal evidence of a range of protein affinities among heparin oligomers. Indeed, when the protein is in short supply (r = 0.2), the most abundant FGF·dp8 complexes contain polyanionic chains incorporating 10 to 11 sulfate groups. However, increasing the relative amount of the protein (to r = 1) shifts this distribution to lower mass, with a maximum corresponding to only nine sulfate groups. It appears that the scarcity of the client protein in solution results in a competition among the heparin oligomers for binding, with the higher charge density species being the winners. When more protein is available, lower charge density heparin oligomers also participate in the binding process. Therefore, despite a wide range of binding-competent dp8 molecules, there is clearly a correlation between the extent of sulfation and the binding affinity. On average, higher sulfation levels favor stronger FGF binding, although some lower charge density species may have high affinity, while certain subsets of higher charge density polyanionic chains are weak binders (vide infra). A very similar trend can be observed from the mass distributions of dp8 chains participating in formation of the FGF2·dp8 complexes: limited availability of protein molecules in solution (r = 0.2) results in preferred utilization of polyanions with the highest charge density (12 sulfate groups), whereas an increased supply of FGF (r = 1) gives rise to equi-abundant complexes, the dp8 components of which incorporate either 11 or 12 sulfate groups. This also provides strong indication of the role of electrostatics in protein/oligoheparin association, although these observations do not necessarily rule out a possibility that there are subsets of (8, 11, 0) species that have very high FGF affinity, exceeding that of some (8, 12, 0) species. A very similar trend is observed in the mass spectra of the FGF/dp10 mixtures with decreasing heparinoid content (Figure 6). No multiple binding to a single chain is observed when the oligoheparin is present at significant molar excess (r = 0.2), and the majority of the FGF·dp10 complexes contain polyanionic chains with relatively high charge density (14 or 15 sulfate groups per oligoheparin chain). Gradual decrease of dp10 content results in utilization of oligoheparins with diminishing charge density in 1:1 complex assembly, as shown by the shift of the red traces in Figure 6 (representing mass distributions of dp10 chains bound to a single protein molecule) towards lower mass. In parallel, complexes of higher stoichiometry become abundant (FGF2·dp10 at r = 0.5 and FGF3·dp10 at r = 4.0). While in each case the average charge density of dp10 chains bridging two protein molecules is always higher than that of the polyanions participating in formation of the FGF·dp10 complexes, the mass distributions also shift towards lower mass as the total level of the oligoheparin in solution decreases. This indicates that the scarcity of the heterogeneous polyanions in solution forces the protein molecules to associate with lower charge density chains regardless of the binding stoichiometry, although accommodation of multiple protein molecules on a single dp10 chain always requires a higher negative charge. These observations demonstrate again the strong correlation between the oligoheparin charge density (i.e., average sulfation level) and its ability to associate with FGF molecule(s).
Insight into Protein Affinity of Heparin Oligomers from the Analysis of Free Polyanions in Solution
Although the analysis of the mass distributions of FGF-bound oligoheparins presented in the preceding section clearly suggests that the average charge density is an important determinant of the binding competence, it leaves open the possibility of a relatively small population of high charge density polyanions being poor FGF binders. Indeed, the data presented in Figures 5 and 6 do not reveal whether all highly sulfated oligoheparin chains are occupied even at very high protein/oligoheparin ratios (e.g., r = 2). To answer this question, we monitored the composition of free (unbound) dp10 chains in solution in the presence of FGF and compared these distributions with that of the dp10 chains in the absence of the protein (Figure 7). This approach aims at observing uneven depletion patterns within the pool of heparinoids following their interaction with the protein. As one can expect, no significant change to the mass distribution of free dp10 chains was observed when they were present in significant (5-fold) molar excess over the protein molecules. However, total depletion of oligoheparins with the highest level of sulfation (15 sulfate groups) is observed even when the dp10 molecules are present at a 2-fold molar excess; significant reduction in abundance of the polyanionic chains with 14 sulfate groups is also evident. This provides unequivocal evidence that all high charge density dp10 chains are strong binders, as they are eliminated from the pool of free oligoheparins even before the FGF/dp10 mixing ratio becomes equimolar. Further increase of the protein/oligoheparin ratio (to r = 1.0) results not only in complete elimination of free high charge density chains (dp10 molecules carrying 14 and 15 sulfate groups), but also a significant reduction of oligoheparins incorporating 13 sulfate groups. Furthermore, the dp10 chains with the lowest degree of sulfation (10 sulfate groups per chain) are represented under these conditions by abundant ionic signal. Since dp10 molecules incorporating 10 sulfate groups were barely detectable in the absence of protein, we conclude that at r = 1.0 there is a significant reduction in the concentration of free heparinoids even with the modest extent of sulfation (≥11), which increases the relative abundance of the (10, 10, x) species and makes their ionic signal prominent in the mass spectrum. Taken together, these observations not only confirm the correlation between binding affinity and oligoheparin sulfation level but also provide unequivocal evidence that there are no weak FGF binders within the subpopulation of polyanions with the highest charge density. This latter conclusion is not trivial, since the dp10 species incorporating 15 sulfate groups may still exhibit enormous structural heterogeneity: the total possible number of isomers within this subpopulation can be calculated using binomial distributions as C15 20 = 15,504 for (10, 15, 0) species and as C15 19 = 3876 for (10, 15, 1) species. The sulfation process is stochastic, but not random, and not all of these isomers are equi-abundant. Nevertheless, a large number of isomers with different sulfation patterns are expected, and the fact that all of them are potent FGF binders provides a strong indication of the major role played by electrostatics in protein/oligoheparin recognition and association.

Mass distributions of free dp10 molecules in solution in the absence (green trace) and in the presence of FGF-1 (r-values are shown on each panel) calculated based on the ionic signals of FGF+2 species in ESI mass spectra. The colored boxes indicate the mass ranges corresponding to the dp10 species with varying levels of sulfation
Elimination of only two sulfate groups from the (10, 15, 0) and (10, 15, 1) species results in a dramatic increase of the possible numbers of isomers (which can be calculated as C13 20 = 77,520 for the (10, 13, 0) species and as C13 19 = 27,132 for the (10, 13, 1) species). Again, even though totally random placement of the sulfate groups is unlikely, the sheer number of possible isomers decreases the probability of having a long contiguous oligoheparin segment with high charge density within the dp10 chain, hence the apparent decrease in the average FGF affinity within the population of oligoheparins carrying 13 sulfate groups. In our interpretation, this lower apparent affinity is a result of the coexistence of several subpopulations: more or less even distribution of the negative charge across the polysaccharide backbone fails to generate strong electrostatic attraction between the positive patch on the protein surface and the oligoheparinoid, whereas concentrating the sulfate groups within a segment of the polysaccharide with physical dimensions similar to the positive patch on the FGF surface results in strong electrostatic attraction and, therefore, higher affinity. Such oligosaccharides are depleted first [reduction of the relative abundance of ions representing the (10, 13, 0) and (10, 13, 1) species is observed at r values as small as 0.5 in Figure 7]. The oligosaccharides with more evenly distributed sulfation are recruited by FGF only at relatively high r (e.g., r = 1.0 in Figure 7) after the higher-affinity polyanions have been already consumed, and the protein is forced to target the so-called “large-capacity but low-affinity sites” [47].
Since HS- or heparin-induced FGF multimerization is the initial step in receptor activation [48], the observed modulation of FGF multimer formation by both protein/oligoheparin ratio and the sulfation patterns (total number of sulfates and their distributions) provides strong support of an HS regulatory role in receptor activation. Unlike classic protein interactions (protein/protein or protein/small ligand), with well-defined binding affinities that can typically be expressed with a single K D value, oligoheparins display a spectrum of affinities towards a given target such as FGF. The protein can then recruit varying populations of the polyanions based not only on their structural characteristics but also on protein/oligoheparin stoichiometry. Such “promiscuity” of oligoheparin/FGF interactions appears to be relevant to signaling processes, as it makes the association of FGF with oligoheparin chains subject to a binding isotherm (similar to the binding isotherm in the pleiotrophin/heparin system reported by Linhardt and co-workers [49]). The probability of multiple GF binding to a single oligoheparin chain increases with r, making the polyanion a template for FGF dimer formation, at least until its saturation. In a sense, oligoheparin binding converts the first step of FGF dimerization to a linear encounter [9]. However, further increase of FGF levels does not necessarily result in a sustained growth of dimer concentration, as the protein must recruit polyanions with progressively lower affinities, akin to generating a negative feedback, thereby preventing excessive (and presumably harmful) receptor over-activation. Metaphorically speaking, the polyanionic chains act as nonlinear amplifiers by enhancing the signal transmitted by growth factors when their concentration is relatively low, and damping the signal at high protein concentration, possibly averting the “overheating” of the molecular signaling circuitry.
Conclusions
The binding of oligoheparins dp8 and dp10 to FGF, a paradigmatic cognate protein for heparan sulftate, provides unequivocal evidence of “promiscuous” complexation driven primarily by electrostatics. Surprisingly, even these relatively short oligoheparins can accommodate up to three protein molecules within a single polyanionic chain. The major determinant of binding efficiency appears to be the overall extent of sulfation controlling not only the affinity towards single proteins but also promoting their multimerization. This process is most notable at elevated protein/heparinoid molar ratios: decreasing the concentration of oligoheparin promotes the binding to lower charge density polyanions. These observations are consistent with glycosaminoglycans in the extracellular matrix acting as non-linier amplifiers of FGF receptor activation: not only do they facilitate FGF dimerization (an obligatory first step in receptor activation) at relatively low protein concentration but also inhibit excess dimer formation at high protein concentration). The present results might support progress in drug delivery and regenerative medicine, including artificial tissue matrices that incorporate heparin to immobilize growth factors. Indeed, our observations suggest that well characterized short heparinoids could modulate growth factor sequestration, in a manner vastly superior to heterogeneous native (intact) heparin.
References
Habuchi, H., Habuchi, O., Kimata, K.: Sulfation pattern in glycosaminoglycan: does it have a code? Glycoconj. J. 21, 47–52 (2004)
Lamanna, W.C., Kalus, I., Padva, M., Baldwin, R.J., Merry, C.L., Dierks, T.: The heparanome—the enigma of encoding and decoding heparan sulfate sulfation. J. Biotechnol. 129, 290–307 (2007)
Lim, J.J., Temenoff, J.S.: The effect of desulfation of chondroitin sulfate on interactions with positively charged growth factors and upregulation of cartilaginous markers in encapsulated MSCs. Biomaterials 34, 5007–5018 (2013)
Gama, C.I., Tully, S.E., Sotogaku, N., Clark, P.M., Rawat, M., Vaidehi, N., Goddard, W.A., Nishi, A., Hsieh-Wilson, L.C.: Sulfation patterns of glycosaminoglycans encode molecular recognition and activity. Nat. Chem. Biol. 2, 467–473 (2006)
Bishop, J.R., Schuksz, M., Esko, J.D.: Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446, 1030–1037 (2007)
Stringer, S.E., Gallagher, J.T.: Heparan sulphate. Int. J. Biochem. Cell Biol. 29, 709–714 (1997)
Seifter, J., Begany, A.J.: Studies on the action of a synthetic heparinoid. Am. J. Med. Sci. 216, 234 (1948)
Ornitz, D.M., Itoh, N.: The fibroblast growth factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 4, 215–266 (2015)
Lander, A.D.: Proteoglycans: master regulators of molecular encounter? Matrix Biol. 17, 465–472 (1998)
Schlessinger, J., Lax, I., Lemmon, M.: Regulation of growth factor activation by proteoglycans: what is the role of the low affinity receptors? Cell 83, 357–360 (1995)
Stickens, D., Zak, B.M., Rougier, N., Esko, J.D., Werb, Z.: Mice deficient in Ext2 lack heparan sulfate and develop exostoses. Development 132, 5055–5068 (2005)
Forsberg, E., Kjellen, L.: Heparan sulfate: lessons from knockout mice. J. Clin. Invest. 108, 175–180 (2001)
Lin, X., Buff, E.M., Perrimon, N., Michelson, A.M.: Heparan sulfate proteoglycans are essential for FGF receptor signaling during Drosophila embryonic development. Development 126, 3715–3723 (1999)
Ashikari-Hada, S., Habuchi, H., Kariya, Y., Itoh, N., Reddi, A.H., Kimata, K.: Characterization of growth factor-binding structures in heparin/heparan sulfate using an octasaccharide library. J. Biol. Chem. 279, 12346–12354 (2004)
Ashikari-Hada, S., Habuchi, H., Sugaya, N., Kobayashi, T., Kimata, K.: Specific inhibition of FGF-2 signaling with 2-O-sulfated octasaccharides of heparan sulfate. Glycobiology 19, 644–654 (2009)
Kayitmazer, A.B., Seeman, D., Minsky, B.B., Dubin, P.L., Xu, Y.: Protein–polyelectrolyte interactions. Soft Matter 9, 2553–2583 (2013)
Olson, S.T., Bjork, I., Sheffer, R., Craig, P.A., Shore, J.D., Choay, J.: Role of the antithrombin-binding pentasaccharide in heparin acceleration of antithrombin-proteinase reactions. Resolution of the antithrombin conformational change contribution to heparin rate enhancement. J. Biol. Chem. 267, 12528–12538 (1992)
Seyrek, E., Dubin, P.: Glycosaminoglycans as polyelectrolytes. Adv. Colloid Interface Sci. 158, 119–129 (2010)
Imberty, A., Lortat-Jacob, H., Pérez, S.: Structural view of glycosaminoglycan–protein interactions. Carbohydr. Res. 342, 430–439 (2007)
Seyrek, E., Dubin, P.L., Tribet, C., Gamble, E.A.: Ionic strength dependence of protein-polyelectrolyte interactions. Biomacromolecules 4, 273–282 (2003)
Kayitmazer, A.B., Quinn, B., Kimura, K., Ryan, G.L., Tate, A.J., Pink, D.A., Dubin, P.L.: Protein specificity of charged sequences in polyanions and heparins. Biomacromolecules 11, 3325–3331 (2010)
Seyrek, E., Dubin, P.L., Henriksen, J.: Nonspecific electrostatic binding characteristics of the heparin-antithrombin interaction. Biopolymers 86, 249–259 (2007)
Catlow, K.R., Deakin, J.A., Wei, Z., Delehedde, M., Fernig, D.G., Gherardi, E., Gallagher, J.T., Pavão M.S., Lyon M.: Interactions of hepatocyte growth factor/scatter factor with various glycosaminoglycans reveal an important interplay between the presence of iduronate and sulfate density. J. Biol. Chem. 283, 5235–5248 (2008)
Kreuger, J., Jemth, P., Sanders-Lindberg, E., Eliahu, L., Ron, D., Basilico, C., Salmivirta, M., Lindahl, U.: Fibroblast growth factors share binding sites in heparan sulphate. Biochem. J. 389, 145–150 (2005)
Jastrebova, N., Vanwildemeersch, M., Rapraeger, A.C., Gimenez-Gallego, G., Lindahl, U., Spillmann, D.: Heparan sulfate-related oligosaccharides in ternary complex formation with fibroblast growth factors 1 and 2 and their receptors. J. Biol. Chem. 281, 26884–26892 (2006)
Jastrebova, N., Vanwildemeersch, M., Lindahl, U., Spillmann, D.: Heparan sulfate domain organization and sulfation modulate FGF-induced cell signaling. J. Biol. Chem. 285, 26842–26851 (2010)
Jones, L.S., Yazzie, B., Middaugh, C.R.: Polyanions and the proteome. Mol. Cell. Proteomics 3, 746–769 (2004)
Venkataraman, G., Shriver, Z., Davis, J.C., Sasisekharan, R.: Fibroblast growth factors 1 and 2 are distinct in oligomerization in the presence of heparin-like glycosaminoglycans. Proc. Natl. Acad. Sci. U. S. A. 96, 1892–1897 (1999)
Bolbach, G.: Matrix-assisted laser desorption/ionization analysis of noncovalent complexes: fundamentals and applications. Curr. Pharm. Des. 11, 2535–2557 (2005)
Pimenova, T., Pereira, C.P., Schaer, D.J., Zenobi, R.: Characterization of high molecular weight multimeric states of human haptoglobin and hemoglobin-based oxygen carriers by high-mass MALDI MS. J. Sep. Sci. 32, 1224–1230 (2009)
Harmer, N.J., Ilag, L.L., Mulloy, B., Pellegrini, L., Robinson, C.V., Blundell, T.L.: Towards a resolution of the stoichiometry of the fibroblast growth factor (FGF)-FGF receptor-heparin complex. J. Mol. Biol. 339, 821–834 (2004)
Crown, S.E., Yu, Y., Sweeney, M.D., Leary, J.A., Handel, T.M.: Heterodimerization of CCR2 chemokines and regulation by glycosaminoglycan binding. J. Biol. Chem. 281, 25438–25446 (2006)
Minsky, B.B., Nguyen, T.V., Peyton, S.R., Kaltashov, I.A., Dubin, P.L.: A heparin decamer bridges a growth factor and an oligolysine by different charge-driven interactions. Biomacromolecules 14, 4091–4098 (2013)
Yu, Y., Sweeney, M.D., Saad, O.M., Crown, S.E., Handel, T.M., Leary, J.A.: Chemokine-glycosaminoglycan binding: specificity for CCR2 ligand binding to highly sulfated oligosaccharides using FT ICR mass spectrometry. J. Biol. Chem. 280, 32200–32208 (2005)
Abzalimov, R.R., Dubin, P.L., Kaltashov, I.A.: Glycosaminoglycans as naturally occurring combinatorial libraries: developing a mass spectrometry-based strategy for characterization of antithrombin interaction with low molecular weight heparin and heparin oligomers. Anal. Chem. 79, 6055–6063 (2007)
Abzalimov, R.R., Kaltashov, I.A.: Electrospray ionization mass spectrometry of highly heterogeneous protein systems: protein ion charge state assignment via incomplete charge reduction. Anal. Chem. 82, 7523–7526 (2010)
Zhao, Y., Abzalimov, R.R., Kaltashov, I.A.: Interactions of intact unfractionated heparin with its client proteins can be probed directly using native electrospray ionization mass spectrometry. Anal. Chem. 88, 1711–1718 (2016)
Kim, J., Blaber, S.I., Blaber, M.: Alternative type I and I′ turn conformations in the beta8/beta9 beta-hairpin of human acidic fibroblast growth factor. Protein Sci. 11, 459–466 (2002)
Rocchia, W., Alexov, E., Honig, B.: Extending the applicability of the nonlinear Poisson-Boltzmann equation: multiple dielectric constants and multivalent ions. J. Phys. Chem. B 105, 6507–6514 (2001)
Rocchia, W., Sridharan, S., Nicholls, A., Alexov, E., Chiabrera, A., Honig, B.: Rapid grid-based construction of the molecular surface and the use of induced surface charge to calculate reaction field energies: applications to the molecular systems and geometric objects. J. Comput. Chem. 23, 128–137 (2002)
Tanford, C., Kirkwood, J.G.: Theory of protein titration curves. I. General equations for impenetrable spheres. J. Am. Chem. Soc. 79, 5333–5339 (1957)
Deakin, J.A., Blaum, B., Gallagher, J.T., Uhrin, D., Lyon, M.: The binding properties of minimal oligosaccharides reveal a common heparan sulfate/dermatan sulfate-binding site in hepatocyte growth factor/scatter factor that can accommodate a wide variety of sulfation patterns. J. Biol. Chem. 284, 6311–6321 (2009)
Mach, H., Volkin, D.B., Burke, C.J., Middaugh, C.R., Linhardt, R.J., Fromm, J.R., Loganathan, D., Mattsson, L.: Nature of the interaction of heparin with acidic fibroblast growth factor. Biochemistry 32, 5480–5489 (1993)
Pavlov, G., Finet, S., Tatarenko, K., Korneeva, E., Ebel, C.: Conformation of heparin studied with macromolecular hydrodynamic methods and X-ray scattering. Eur. Biophys. J. 32, 437–449 (2003)
Casu, B., Petitou, M., Provasoli, M., Sinay, P.: Conformational flexibility: a new concept for explaining binding and biological properties of iduronic acid-containing glycosaminoglycans. Trends Biochem. Sci. 13, 221–225 (1988)
Henriksen, J., Ringborg, L.H., Roepstorrf, P.: On-line size-exclusion chromatography/mass spectrometry of low molecular mass heparin. J. Mass Spectrom. 39, 1305–1312 (2004)
Nugent, M.A., Zaia, J., Spencer, J.L.: Heparan sulfate-protein binding specificity. Biochemistry [Biokhimiia] 78, 726–735 (2013)
Spivak-Kroizman, T., Lemmon, M.A., Dikic, I., Ladbury, J.E., Pinchasi, D., Huang, J., Jaye, M., Crumley, G., Schlessinger, J., Lax, I.: Heparin-induced oligomerization of FGF molecules is responsible for FGF receptor dimerization, activation, and cell proliferation. Cell 79, 1015–1024 (1994)
Fath, M., VanderNoot, V., Kilpelainen, I., Kinnunen, T., Rauvala, H., Linhardt, R.J.: Interaction of soluble and surface-bound heparin binding growth-associated molecule with heparin. FEBS Lett. 454, 105–108 (1999)
Acknowledgements
This work was supported in part by a grant from the National Institutes of Health R01 GM112666. The authors thank Professor Robert J. Linhardt (Rensselaer Polytechnic Institute, Troy, NY) for providing the FGF-1 sample, and Dr. Rinat R. Abzalimov (UMass-Amherst; presently at CUNY Advanced Science Research Center, Harlem, NY) for help with the experimental work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Minsky, B.B., Dubin, P.L. & Kaltashov, I.A. Electrostatic Forces as Dominant Interactions Between Proteins and Polyanions: an ESI MS Study of Fibroblast Growth Factor Binding to Heparin Oligomers. J. Am. Soc. Mass Spectrom. 28, 758–767 (2017). https://doi.org/10.1007/s13361-017-1596-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-017-1596-0