
Abstract
In the last two decades, computational tools for mass spectrometry-based proteomics data analysis have evolved from a few stand-alone software solutions serving specific goals, such as the identification of amino acid sequences based on mass spectrometry spectra, to large-scale complex pipelines integrating multiple computer programs to solve a collection of problems. This software evolution has been mostly driven by the appearance of novel technologies that allowed the community to tackle complex biological problems, such as the identification of proteins that are differentially expressed in two samples under different conditions. The achievement of such objectives requires a large suite of programs to analyze the intricate mass spectrometry data. Our laboratory addresses complex proteomics questions by producing and using algorithms and software packages. Our current computational pipeline includes, among other things, tools for mass spectrometry raw data processing, peptide and protein identification and quantification, post-translational modification analysis, and protein functional enrichment analysis. In this paper, we describe a suite of software packages we have developed to process mass spectrometry-based proteomics data and we highlight some of the new features of previously published programs as well as tools currently under development.

ᅟ
Similar content being viewed by others


Introduction
Computer science and bioinformatics played a crucial role in the development of the field of mass spectrometry (MS)-based proteomics. As MS instruments evolved, the data generated grew increasingly intricate. In response, computational strategies were developed to dramatically increase our capacity to extract knowledge from MS data. Foremost among these is the SEQUEST algorithm [1] that allowed the high-throughput identification of thousands of tandem MS (MS/MS) spectra in a reasonable time; such a feat was unthinkable 20 years ago. Since then, multiple peptide and protein sequence database search engines have been proposed (Mascot [2], OMSSA [3], X!Tandem [4], MS-GF [5], Andromeda [6], and, more recently, Morpheus [7]). However, protein identification still remains an important challenge, and MS-based proteomics now involves a larger variety of computational and data analysis problems.
MS now allows, among other things, the identification and quantification of post-translationally modified proteins as well as interacting proteins and the differential quantification of proteins in samples under different experimental conditions [8–11]. These analyses are achieved with a large collection of software packages for processing MS data and for performing statistical analyses. A large number of MS-based proteomics analyses can be typically broken down into three initial steps: first, the raw data produced by mass spectrometers need to be processed and transformed into a suitable input for programs performing data analysis. Second, the spectra are matched to peptides using protein sequence database searching, spectral library searching, or de novo sequencing. Third, the statistical significance of peptide and protein identifications needs to be assessed. Subsequent steps may vary based on the goals of the experiment. A variety of computational tools may be used to quantify peptides and proteins (e.g., ProRata [12], pQuant [13], and MaxQuant [14]), as well as identify PTMs (e.g., Ascore [15], InsPecT [16]). Of note, the three previously enumerated initial steps may differ in MS-based proteomics workflows that involve methods such as selected reaction monitoring (SRM) and data-independent acquisition (DIA) [17, 18]. There is also a collection of publicly available software packages that can be used to filter out nonspecific protein–protein interactions (PPIs) and identify those of high confidence in datasets produced using affinity purification coupled to MS. While we do not explicitly describe these tools here, we count among them software packages such as SAINT [19], Decontaminator [20, 21], Mist [22], and Compass [23].
The large number of complex software packages required to perform the computational analysis transforming the mass spectrometer raw data into meaningful proteomics and biological discoveries can make the processing of large-scale datasets quite challenging, especially when relying on multiple applications from different sources. Since the output of a given program often serves as input for another computational tool, paired outputs and inputs have to remain compatible over time from a file format and conceptual perspective throughout the computational pipeline. A number of computational proteomics pipelines that include the complete set of tools necessary to perform all or a subset of the analysis of an MS-based proteomics experiment have been proposed. Some of these pipelines present a comprehensive software solution for MS-based proteomics data analysis and include among their modules computational tools for peptide and protein identification and quantification (e.g., MaxQuant [14], pFind Studio [24], PEAKS [25], OpenMS Proteomics Pipeline (TOPP) [26], and Trans-Proteomic Pipeline (TPP) [27]). Others focus on the quantitative analysis and biological interpretation of MS-based proteomics data (e.g., PatternLab [28]) or on the protein identification and the statistical validation of the results (e.g., Mascot-Percolator interface package [29]). In addition, a set of modular open-source cross-platform computational tools and libraries processing and analyzing MS data are available under the ProteoWizard software project [30]. ProteoWizard includes among other things the Skyline software package [31] that allows the analysis of quantitative MS-based proteomics data acquired using a variety of methods such as SRM and DIA.
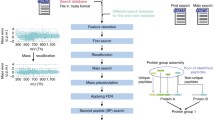
Our group has also developed a collection of computational tools to perform the computational analysis of MS-based proteomics datasets from the raw data to its biological interpretation [1, 32–35]. Our tools were developed using a set of requirements (of which we have a complete control) in order to ensure the compatibility of each tool throughout the evolution of our pipeline. Figure 1 illustrates our main computational pipeline. While all of the tools developed by our group are publically available as stand-alone applications (http://fields.scripps.edu/researchtools.php), a large number of these software packages are also available in a web-based comprehensive environment, called Integrated Proteomics Pipeline (IP2) (Integrated Proteomics Applications) that allows the analysis of large-scale MS-based proteomics datasets. While some users prefer executing each computational tool in the pipeline independently, IP2 facilitates the analysis of large-scale complex datasets by providing a single interface and work environment for the implementation of the complete analysis pipeline. In this article, we discuss numerous tools that compose our computational pipeline for MS-based proteomics analysis, with an emphasis on some of their new features. We also present our most recent software packages and the algorithms that are currently under development in our laboratory.

Schematic representation of the main computational pipeline of our laboratory. For a given experiment, the computational tools in the large black box may be used based on the goals of the experiment. Green boxes represent software packages developed in our laboratory and yellow boxes illustrate those designed by third parties
Computational Pipeline
Mass Spectrometry Raw Data Processing
The first step of the vast majority of MS-based proteomics experiments still remains peak extraction from MS raw data. Our laboratory previously developed a tool named RawExtract that converts binary Thermo Fisher Scientific RAW files to text-based files in MS1/MS2 format [36], which can be used for peptide identification. RawExtract has evolved over time and its latest version, named RawConverter, introduces a set of novel features. RawConverter can convert RAW files to MS1/MS2, but also to MGF (Mascot Generic Format), or mzXML [37] formats. It also now provides file format conversions from mzXML and mzML files [38] to MS1/MS2 and MGF files, and from MGF files to MS2 files. These conversions conveniently fulfill the MS file format requirements of the vast majority of downstream data analysis tools that are publically available, including those of our computational pipeline.
In addition to extracting the MS or MS/MS data from RAW files generated by Thermo Fisher Scientific instruments, RawConverter addresses problems regarding the selection of the monoisotopic peaks of precursor ions. The inaccurate precursor mass assignment of an MS/MS spectrum significantly increases the probability of introducing false positives in subsequent peptide and protein identifications. Mass spectrometers from certain manufacturers tend to label the peak with the highest intensity in a given isolation window as the precursor peak when spectra are collected using data-dependent acquisition (DDA). Generally, this precursor peak can be considered as the monoisotopic peak for short peptides (with mass equal to or less than 1,500 Da) since their monoisotopic peaks typically have the highest intensities [39]. However, the monoisotopic ions of longer peptides (with mass greater than 1,500 Da) may not be those with the highest intensities [39], making the above straightforward peak picking strategy far from ideal for large peptides. Existing tools available in the ProteoWizard software project [30] and pParse [40] have recently made progress on tackling this issue. The RawConverter algorithm also addresses this problem by combining the Averagine model [41] and the information of successive MS1 scans to select the monoisotopic peak for each precursor ion.
Another problem arises in the context of DIA methods. Most MS instruments do not supply precursor m/z and charge information of the peptides in a given wide acquisition window; they merely provide the middle m/z and the size of the isolation windows [18]. Providing more accurate precursor information to database search engines would allow a reduction of the search space and, therefore, an increased discriminative power when scoring peptide-spectrum matches (PSMs). This would benefit the majority of the current tools assessing the confidence of protein and peptide identifications [32, 42, 43]. RawConverter attempts to address this issue by enumerating and evaluating all possible precursor isotopic envelopes in a given DIA isolation window. Users can define the maximum number of precursor isotopic envelopes reported. The most confident envelopes are selected to determine the possible precursor m/z values and charge states. MS/MS spectra can then be duplicated for each possible precursor, thereby allowing each spectra to be individually searched for peptide identification.
Peptide and Protein Identification and Statistical Significance Assessment
Once extracted, the MS data output by RawConverter (MS2 file format) is searched by our protein sequence database search engine ProLuCID [44] to identify PSMs. ProLuCID, which is implemented as a platform-independent Java program, is inspired by the SEQUEST algorithm and, therefore, uses a modified cross-correlation (XCorr) calculation to score a spectrum against the theoretical spectrum of a given peptide sequence. ProLuCID supports low- and high-resolution MS data. It also introduces the computation of a binomial probability serving as a preliminary score to pre-filter spectra and to improve computational performances. In addition, it reports a Z-score for the PSM with the highest XCorr for each spectrum, which can be used to filter search results.
The significance of the set of PSMs produced by most database search engines needs to be statistically assessed by computational tools, such as Percolator [42]. In our pipeline, PSMs found with ProLuCID are statistically assessed using DTASelect [32, 43]. DTASelect also identifies the set of proteins present in the analyzed sample based on ProLuCID’s PSMs. Over the years, DTASelect’s PSM filtering accuracy has been improved. Furthermore, the latest version (DTASelect 2) now outputs the set of spectra, peptides, or proteins identified under a user-selected false discovery rate for a given experiment. Thanks to recent improvements, DTASelect 2 provides a great filtering flexibility by allowing users to define up to 150 different parameters. For example, a filter can be set so that the only reported proteins are those identified by a certain number of peptides and that at least one of these peptides obtained a XCorr above a given threshold. These new filters allow the generation of high-confidence datasets by increasing the number of protein identifications under a given false discovery rate.
While the vast majority of the samples analyzed by MS/MS in our laboratory involve organisms for which most protein sequences are known, we sometimes process samples originating from organisms for which the genomes are not yet sequenced. This renders a typical sequence database search impossible. For such samples, peptides are usually identified using the de novo sequencing software package pNovo [45].
Peptide and Protein Quantification
Peptide and protein quantification is the next step of the data analysis in several MS-based proteomics pipelines. Computational tools such as ProRata [12], pQuant [13], and MaxQuant [14] have been proposed to calculate accurate proteomics quantitative measurements. We have also previously introduced a software tool named Census [33]. Census is capable of using the vast majority of quantitative MS-based proteomics strategies and can take as its input DTASelect’s output as well as pepXML and mzXML files. Census can quantify peptides and proteins labeled using a variety of labeling strategies (e.g., 15N [46], SILAC [47], iTRAQ [48], TMT [49], dimethyl [50, 51], 18O [52]), as well as label-free strategies for both high- and low-resolution MS data [spectral counting and extracted-ion chromatogram (XIC)-based quantification] (see Figure 2). Census can be used to analyze large-scale quantitative datasets and, when coupled to high-resolution MS data, shows improved quantification efficiency [33]. Among its numerous features, Census quantifies peptides originating from DIA data with high sensitivity by filtering out noisy fragment ions and also builds chromatograms from peak lists to quantify stable isotope-labeled peptides.

Main MS-based quantitative proteomics approaches supported by Census
The recently released Census ver. 2 [53] comprises novel features for the analysis of peptides quantified using tandem mass tag (TMT) reporter ions. Included among these are a reporter ion impurity correction, a reporter ion minimum intensity threshold filter, and an optional weighted normalization that corrects mixing errors. Census 2 can also process MS experiments performed using HCD, CID/HCD double-play, HCD MS3, or MultiNotch MS3 [54, 55] data. Additional features have also been included for quantification using metabolic labeling (e.g., 15N, SILAC). Among these are the calculation of a reverse ratio for the label swap experimental setup [56, 57] and the assessment of the statistical significance of differentially expressed peptides. More recently, improvements were implemented in Census 2 for the calculation of 15N enrichment ratios. Census 2 uses the elemental composition of amino acids to calculate the isotopic distributions of 15N enriched peptides. As 15N labeling shifts the mass of peptides based on the number of nitrogen atoms they contain, Census 2 uses all possible theoretical isotope distributions and maps them to the experimental ones to find the best match using a linear regression. Census 2 computes the atomic enrichment for each peptide independently, as it can vary based on a given protein’s turnover rate.
Post-Translational Modifications Discovery and Assessment
While ProLuCID’s database search algorithm allows the identification of post-translationally modified peptides, some proteomics studies might require a more in-depth analysis of certain PTM events. Hence, our computational pipeline also includes the probability-based phosphorylation localization tool Ascore, which was developed by Beausoleil et al. [15]. In addition, we developed an algorithm named Debunker [34] that uses a support vector machine binary classifier to assess the phosphorylation status of a given peptide. Debunker may be used in combination with Ascore to assess the confidence of phosphorylation status and site localization for a given peptide.
Functional Enrichment Analysis
Functional enrichment analyses are often used to generate hypotheses regarding the underlying mechanisms revealed in MS-based proteomics datasets. Such analyses can consist of identifying gene ontology (GO) [58] annotations that are over-represented in a set of differentially expressed proteins or finding biological pathways that are significantly implicated in a set of protein–protein interactions (PPIs). Our computational pipeline currently includes the use of the Ingenuity Pathway Analysis (IPA, QIAGEN Redwood City, CA, USA www.qiagen.com/ingenuity) and of DAVID [59] as well as Ontologizer [60] for GO enrichment analyses.
We also recently developed our own functional enrichment analysis algorithm for MS-based proteomics quantification experiments (PSEA-Quant [35]). Enrichment analyses of gene/protein sets, such as those obtained from the GO and the Molecular Signature databases [61] were often developed for genomic datasets [62] and are poorly adapted to some of the particularities of MS-based proteomics data, including the level of reproducibility of MS quantitative measurements. They also tend to require the use of arbitrary thresholds to define the subset of proteins in a dataset on which the enrichment analysis should be performed. PSEA-Quant addresses both of these problems. This web-based user-friendly algorithm, inspired by GSEA [62], entails a novel protein set enrichment analysis for label-free and labeled MS-based quantitative proteomics. Unlike GSEA, PSEA-Quant allows the analysis of proteomics samples originating from single or multiple conditions. This Java program uses Census’ output, while supporting other file formats, to identify protein sets that are statistically enriched significantly among abundant proteins that are quantified with high reproducibility across a set of replicates. PSEA-Quant was used to highlight putative underlying mechanisms of cystic fibrosis [35], among other things.
Algorithms and Tools Currently in Development
This article presents a snapshot of the state of our current computational pipeline. However, our suite of software packages is under constant evolution. It continuously adapts to novel technologies with the goal of producing high impact discoveries. For instance, we are currently developing algorithms for the identification of cross-linked peptides. In addition, we are designing a novel blind search algorithm that allows the identification of peptides with any unspecified PTMs or amino acid substitutions using MS/MS spectra. We also continue to improve our intact protein (top-down) computational analysis pipeline, which includes tools such as ProSightPC (Thermo Fisher Scientific) and ByOnic [63], by developing algorithms that improve protein identification sensitivity and estimate the number of protein species in a dataset analyzed through top-down MS [64].
A recent addition to our computational pipeline is a tool named PrOntoNet, which infers PPIs from a list of identified proteins in organisms for which there are very few known PPIs in the literature and for which the proteins are largely uncharacterized functionally. PrOntoNet uses the output of DTASelect to recover known PPIs from identified proteins in the STRING [65] and BioGRID [66] databases. It then uses blast2GO [67] to associate GO annotations to the proteins in the dataset. PrOntoNet finally uses the proportion of overlap between the GO annotations of all protein pairs in the dataset in order to infer interactions between them.
The tools described above provide a large amount of information on the PSMs and the proteins identified in a given proteomics dataset. The results from several bioinformatics analyses and data from public external resources and databases are typically processed and combined in order to convey to the scientific community a comprehensive and meaningful message about a proteomics study. This task often involves the manual integration of in-house software and script results, as well as data from spreadsheets, into customized summary tables that are later published in an article. The large-scale datasets and results produced with MS-based proteomics pose important challenges in terms of long-term storage and data reusability. Reproducing the creation process of complex summary tables can be difficult after a long period of time, especially when attempted by someone other than the original author. Databases such as PRIDE [68], MassIVE (massive.ucsd.edu), and PeptideAtlas [69], which are all included in the ProteomeXchange consortium [70], allow the long-term storage and distribution of data. However, for the most part they do not permit the full integration of custom computational and statistical significance results. We are currently tackling both of these challenges simultaneously by designing a software package named Proteomics INTegrator (PINT). PINT is an open-source platform-independent Java program built on top of a MySQL database engine that provides an environment to store data and integrate results from various computational analyses (including ProLuCID, DTASelect, and Census), which may originate from different proteomics approaches. The protein annotations available in the UniProtKB repository [71] are automatically appended to all datasets stored in PINT. PINT allows users to easily import, visualize, and download data though a user-friendly web interface, thereby facilitating its dissemination and accessibility. In addition, our tool also provides a powerful and flexible query system allowing the retrieval of specific elements and values (e.g., quantification measurements, confidence scores, statistical significance results, manual annotations, UniprotKB protein annotations) in the data of a given experiment or across several experiments simultaneously.
Conclusion
Our computational pipeline has come a long way since the publication of the SEQUEST algorithm. We strive to continuously produce and use novel algorithms that take advantage of new MS technological improvements. We strongly believe that computational approaches that aim to make the most out of cutting-edge technologies will continue to yield high impact biological discoveries.
References
Eng, J.K., McCormack, A.L., Yates III, J.R.: An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass. Spectrom. 5, 976–989 (1994)
Cottrell, J.S., London, U.: Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 (1999)
Geer, L.Y., Markey, S.P., Kowalak, J.A., Wagner, L., Xu, M., Maynard, D.M., Yang, X., Shi, W., Bryant, S.H.: Open mass spectrometry search algorithm. J. Proteome. Res. 3, 958–964 (2004)
Craig, R., Beavis, R.C.: TANDEM: matching proteins with tandem mass spectra. Bioinformat. 20(9), 1466–1467 (2004)
Kim, S., Gupta, N., Pevzner, P.A.: Spectral probabilities and generating functions of tandem mass spectra: a strike against decoy databases. J. Proteome. Res. 7, 3354–3363 (2008)
Cox, J., Neuhauser, N., Michalski, A., Scheltema, R.A., Olsen, J.V., Mann, M.: Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome. Res. 10, 1794–1805 (2011)
Wenger, C.D., Coon, J.J.: A proteomics search algorithm specifically designed for high-resolution tandem mass spectra. J. Proteome. Res. 12, 1377–1386 (2013)
Yates, J.R., Ruse, C.I., Nakorchevsky, A.: Proteomics by mass spectrometry: approaches, advances, and applications. Annu. Rev. Biomed. Eng. 11, 49–79 (2009)
Gingras, A.-C., Gstaiger, M., Raught, B., Aebersold, R.: Analysis of protein complexes using mass spectrometry. Nat. Rev. Mol. Cell. Biol. 8, 645–654 (2007)
Sinz, A.: Chemical cross-linking and mass spectrometry to map three-dimensional protein structures and protein–protein interactions. Mass. Spectrom. Rev. 25, 663–682 (2006)
Bantscheff, M., Schirle, M., Sweetman, G., Rick, J., Kuster, B.: Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem. 389, 1017–1031 (2007)
Pan, C., Kora, G., McDonald, W.H., Tabb, D.L., VerBerkmoes, N.C., Hurst, G.B., Pelletier, D.A., Samatova, N.F., Hettich, R.L.: ProRata: a quantitative proteomics program for accurate protein abundance ratio estimation with confidence interval evaluation. Anal. Chem. 78, 7121–7131 (2006)
Liu, C., Song, C.-Q., Yuan, Z.-F., Fu, Y., Chi, H., Wang, L., Fan, S.-B., Zhang, K., Zeng, W.-F., He, S.-M., Dong, M.-Q., Sun, R.-X.: pQuant improves quantitation by keeping out interfering signals and evaluates the accuracy of calculated ratios. Anal. Chem. 86(11), 5286–5294 (2014)
Cox, J., Mann, M.: MaxQuant enables high peptide identification rates, individualized ppb-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008)
Beausoleil, S.A., Villén, J., Gerber, S.A., Rush, J., Gygi, S.P.: A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 (2006)
Tanner, S., Shu, H., Frank, A., Wang, L.-C., Zandi, E., Mumby, M., Pevzner, P.A., Bafna, V.: InsPecT: identification of post-translationally modified peptides from tandem mass spectra. Anal. Chem. 77, 4626–4639 (2005)
Gillet, L.C., Navarro, P., Tate, S., Röst, H., Selevsek, N., Reiter, L., Bonner, R., Aebersold, R.: Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteom. 11, O111.016717 (2012)
Venable, J.D., Dong, M.-Q., Wohlschlegel, J., Dillin, A., Ates, J.R.: Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat. Methods 1, 39–45 (2004)
Choi, H., Larsen, B., Lin, Z.-Y., Breitkreutz, A., Mellacheruvu, D., Fermin, D., Qin, Z.S., Tyers, M., Gingras, A.-C., Nesvizhskii, A.I.: SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods 8, 70–73 (2011)
Lavallée-Adam, M., Cloutier, P., Coulombe, B., Blanchette, M.: Modeling contaminants in AP-MS/MS experiments. J. Proteome. Res. 10, 886–895 (2010)
Lavallée-Adam, M., Rousseau, J., Domecq, C., Bouchard, A., Forget, D., Faubert, D., Blanchette, M., Coulombe, B.: Discovery of cell compartment specific protein–protein interactions using affinity purification combined with tandem mass spectrometry. J. Proteome. Res. 12, 272–281 (2012)
Jäger, S., Cimermancic, P., Gulbahce, N., Johnson, J.R., McGovern, K.E., Clarke, S.C., Shales, M., Mercenne, G., Pache, L., Li, K., Hernandez, H., Jang, G.M., Roth, S.L., Akiva, E., Marlett, J., Stephens, M., D'Orso, I., Fernandes, J., Fahey, M., Mahon, C., O'Donogue, A.J., Todorovic, A., Morris, J.H., Maltby, D.A., Alber, T., Cagney, G., Bushman, F.D., Young, J.A., Chanda, S.K., Sundquist, W.I., Kortemme, T., Hernandez, R.D., Craik, C.S., Burlingame, A., Sali, A., Frankel, A.D., Krogan, N.J.: Global landscape of HIV-human protein complexes. Nature 481, 365–370 (2012)
Sowa, M.E., Bennett, E.J., Gygi, S.P., Harper, J.W.: Defining the human deubiquitinating enzyme interaction landscape. Cell. 138, 389–403 (2009)
Li, D., Fu, Y., Sun, R., Ling, C.X., Wei, Y., Zhou, H., Zeng, R., Yang, Q., He, S., Gao, W.: pFind: a novel database-searching software system for automated peptide and protein identification via tandem mass spectrometry. Bioinformatics 21, 3049–3050 (2005)
Ma, B., Zhang, K., Hendrie, C., Liang, C., Li, M., Doherty-Kirby, A., Lajoie, G.: PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid. Commun. Mass. Spectrom. 17, 2337–2342 (2003)
Kohlbacher, O., Reinert, K., Gröpl, C., Lange, E., Pfeifer, N., Schulz-Trieglaff, O., Sturm, M.: TOPP—the OpenMS proteomics pipeline. Bioinformatics 23, e191–e197 (2007)
Keller, A., Eng, J., Zhang, N., Li, X., Aebersold, R.: A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol. Syst. Biol. 1, 2005.0017 (2005)
Carvalho, P.C., Fischer, J.S.G., Chen, E.I., Yates III, J.R., Barbosa, V.C.: PatternLab for proteomics: a tool for differential shotgun proteomics. BMC. Bioinformat. 9, 316 (2008)
Brosch, M., Yu, L., Hubbard, T., Choudhary, J.: Accurate and sensitive peptide identification with Mascot Percolator. J. Proteome. Res. 8, 3176–3181 (2009)
Kessner, D., Chambers, M., Burke, R., Agus, D., Mallick, P.: ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536 (2008)
MacLean, B., Tomazela, D.M., Shulman, N., Chambers, M., Finney, G.L., Frewen, B., Kern, R., Tabb, D.L., Liebler, D.C., MacCoss, M.J.: Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 (2010)
Tabb, D.L., McDonald, W.H., Yates, J.R.: DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome. Res. 1, 21–26 (2002)
Park, S.K., Venable, J.D., Xu, T., Yates, J.R.: A quantitative analysis software tool for mass spectrometry-based proteomics. Nat. Methods 5, 319–322 (2008)
Lu, B., Ruse, C., Xu, T., Park, S.K., Yates, J.: Automatic validation of phosphopeptide identifications from tandem mass spectra. Anal. Chem. 79, 1301–1310 (2007)
Lavallée-Adam, M., Rauniyar, N., McClatchy, D.B., Yates J.R. III: PSEA-Quant: a protein set enrichment analysis on label-free and label-based protein quantification data. J. Proteome Res. 13(12), 5496–5509 (2014)
McDonald, W.H., Tabb, D.L., Sadygov, R.G., MacCoss, M.J., Venable, J., Graumann, J., Johnson, J.R., Cociorva, D., Yates, J.R.: MS1, MS2, and SQT—three unified, compact, and easily parsed file formats for the storage of shotgun proteomic spectra and identifications. Rapid Commun. Mass. Spectrom. 18, 2162–2168 (2004)
Pedrioli, P.G.A., Eng, J.K., Hubley, R., Vogelzang, M., Deutsch, E.W., Raught, B., Pratt, B., Nilsson, E., Angeletti, R.H., Apweiler, R., Cheung, K., Costello, C.E., Hermjakob, H., Huang, S., Julian, R.K., Kapp, E., McComb, M.E., Oliver, S.G., Omenn, G., Paton, N.W., Simpson, R., Smith, R., Taylor, C.F., Zhu, W., Aebersold, R.: A common open representation of mass spectrometry data and its application to proteomics research. Nat. Biotechnol. 22, 1459–1466 (2004)
Deutsch, E.: mzML: a single, unifying data format for mass spectrometer output. Proteomics 8, 2776–2777 (2008)
Nassar, A.F., Hollenberg, P., Scatina, J.: Drug Metabolism Handbook: Concepts and Applications. John Wiley and Sons, Hoboken, NJ, p. 216 (2009)
Liu, C., Wang, H.-P., Sun, R.-X., Fu, Y., Zhang, J.-F., Wang, L.-H., Chi, H., Li, Y., Xiu, L.-Y., Wang, W.-P., He, S.M.: pParse: a method for accurate determination of monoisotopic peaks in high-resolution mass spectra. Proteomics 12, 226–235 (2012)
Senko, M.W., Beu, S.C., McLafferty, F.W.: Determination of monoisotopic masses and ion populations for large biomolecules from resolved isotopic distributions. J. Am. Soc. Mass. Spectrom 6, 229–233 (1995)
Käll, L., Canterbury, J.D., Weston, J., Noble, W.S., MacCoss, M.J.: Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923–925 (2007)
Cociorva, D., L Tabb, D., Yates, J.R.: Validation of tandem mass spectrometry database search results using DTASelect. Curr. Protoc. Bioinformat. 16, 13.4.1–13.4.14 (2007)
Xu, T., Venable, J.D., Park, S.K., Cociorva, D., Lu, B., Liao, L., Wohlschlegel, J., Hewel, J., Yates III, J.R.: ProLuCID, a fast and sensitive tandem mass spectra-based protein identification program. Mol. Cell. Proteom. 5, S174–S174 (2006)
Chi, H., Sun, R.-X., Yang, B., Song, C.-Q., Wang, L.-H., Liu, C., Fu, Y., Yuan, Z.-F., Wang, H.-P., He, S.-M., Dong, M.-Q.: pNovo: de novo peptide sequencing and identification using HCD spectra. J. Proteome. Res. 9 III, 2713–2724 (2010)
Washburn, M.P., Ulaszek, R., Deciu, C., Schieltz, D.M., Yates, J.R.: Analysis of quantitative proteomic data generated via multidimensional protein identification technology. Anal. Chem. 74 III, 1650–1657 (2002)
Ong, S.-E., Blagoev, B., Kratchmarova, I., Kristensen, D.B., Steen, H., Pandey, A., Mann, M.: Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom. 1, 376–386 (2002)
Ross, P., Huang, Y.N., Marchese, J.N., Williamson, B., Parker, K., Hattan, S., Khainovski, N., Pillai, S., Dey, S., Daniels, S., Purkayastha, S., Juhasz, P., Martin, S., Bartlet-Jones, M., He, F., Jacobson, A., Pappin, D.J.: Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteom. 3, 1154–1169 (2004)
Thompson, A., Schäfer, J., Kuhn, K., Kienle, S., Schwarz, J., Schmidt, G., Neumann, T., Hamon, C.: Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 75, 1895–1904 (2003)
Hsu, J.-L., Huang, S.-Y., Chow, N.-H., Chen, S.-H.: Stable-isotope dimethyl labeling for quantitative proteomics. Anal. Chem. 75, 6843–6852 (2003)
Boersema, P.J., Raijmakers, R., Lemeer, S., Mohammed, S., Heck, A.J.R.: Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat. Protoc. 4, 484–494 (2009)
Yao, X., Freas, A., Ramirez, J., Demirev, P.A., Fenselau, C.: Proteolytic 18O labeling for comparative proteomics: model studies with two serotypes of adenovirus. Anal. Chem. 73, 2836–2842 (2001)
Park, S.K.R., Aslanian, A., McClatchy, D.B., Han, X., Shah, H., Singh, M., Rauniyar, N., Moresco, J.J., Pinto, A.F.M., Diedrich, J.K., Delahunty, C., Yates J.R. III: Census 2: isobaric labeling data analysis. Bioinformatics. 30(15), 2208–2209 (2014)
Ting, L., Rad, R., Gygi, S.P., Haas, W.: MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 8, 937–940 (2011)
McAlister, G.C., Nusinow, D.P., Jedrychowski, M.P., Wühr, M., Huttlin, E.L., Erickson, B.K., Rad, R., Haas, W., Gygi, S.P.: MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 86, 7150–7158 (2014)
Cox, J., Mann, M.: Quantitative, high-resolution proteomics for data-driven systems biology. Annu. Rev. Biochem. 80, 273–299 (2011)
Park, S.-S., Wu, W.W., Zhou, Y., Shen, R.-F., Martin, B., Maudsley, S.: Effective correction of experimental errors in quantitative proteomics using stable isotope labeling by amino acids in cell culture (SILAC). J. Proteom. 75, 3720–3732 (2012)
Ashburner, M., Ball, C.A., Blake, J.A., Botstein, D., Butler, H., Cherry, J.M., Davis, A.P., Dolinski, K., Dwight, S.S., Eppig, J.T., Harris, M.A., Hill, D.P., Issel-Tarver, L., Kasarskis, A., Lewis, S., Matese, J.C., Richardson, J.E., Ringwald, M., Rubin, G.M., Sherlock, G.: Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000)
Dennis Jr., G., Sherman, B.T., Hosack, D.A., Yang, J., Gao, W., Lane, H.C., Lempicki, R.A.: DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 4, P3 (2003)
Bauer, S., Grossmann, S., Vingron, M., Robinson, P.N.: Ontologizer 2.0—a multifunctional tool for GO term enrichment analysis and data exploration. Bioinformatics 24, 1650–1651 (2008)
Liberzon, A., Subramanian, A., Pinchback, R., Thorvaldsdóttir, H., Tamayo, P., Mesirov, J.: Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011)
Subramanian, A., Tamayo, P., Mootha, V.K., Mukherjee, S., Ebert, B.L., Gillette, M.A., Paulovich, A., Pomeroy, S.L., Golub, T.R., Lander, E.S., Mesirov, J.: Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102, 15545–15550 (2005)
Bern, M., Cai, Y., Goldberg, D.: Lookup peaks: a hybrid of de novo sequencing and database search for protein identification by tandem mass spectrometry. Anal. Chem. 79, 1393–1400 (2007)
Han, X., Wang, Y., Aslanian, A., Bern, M., Lavallée-Adam, M., Yates III, J.R.: Sheathless capillary electrophoresis-tandem mass spectrometry for top-down characterization of pyrococcus furiosus proteins on a proteome scale. Anal. Chem. 86, 11006–11012 (2014)
Szklarczyk, D., Franceschini, A., Kuhn, M., Simonovic, M., Roth, A., Minguez, P., Doerks, T., Stark, M., Muller, J., Bork, P., Jensen, L.J., von Mering, C.: The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 39, D561–D568 (2011)
Stark, C., Breitkreutz, B.J., Reguly, T., Boucher, L., Breitkreutz, A., Tyers, M.: BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 34, D535 (2006)
Conesa, A., Götz, S., García-Gómez, J.M., Terol, J., Talón, M., Robles, M.: Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005)
Martens, L., Hermjakob, H., Jones, P., Adamski, M., Taylor, C., States, D., Gevaert, K., Vandekerckhove, J., Apweiler, R.: PRIDE: the proteomics identifications database. Proteomics 5, 3537–3545 (2005)
Deutsch, E.W., Eng, J.K., Zhang, H., King, N.L., Nesvizhskii, A.I., Lin, B., Lee, H., Yi, E.C., Ossola, R., Aebersold, R.: Human plasma peptideatlas. Proteomics 5, 3497–3500 (2005)
Vizcaíno, J.A., Deutsch, E.W., Wang, R., Csordas, A., Reisinger, F., Ríos, D., Dianes, J.A., Sun, Z., Farrah, T., Bandeira, N., Binz, P.A., Xenarios, I., Eisenacher, M., Mayer, G., Gatto, L., Campos, A., Chalkley, R.J., Kraus, H.J., Albar, J.P., Martinez-Bartolomé, S., Apweiler, R., Omenn, G.S., Martens, L., Jones, A.R., Hermjakob, H.: ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 (2014)
UniProt Consortium: Activities at the universal protein resource (UniProt). Nucleic Acids Res. 42.D1, D191–D198 (2014)
Acknowledgments
The authors are grateful to Claire M. Delahunty for helpful discussions and comments. They acknowledge funding from the following National Institute of Health grants: P41 GM103533, R01 MH067880, R01 MH100175, UCLA/NHLBI Proteomics Centers (HHSN268201000035C), and 1U54GM114833. M.L.A. holds a postdoctoral fellowship from the Fonds de recherche du Québec – nature et technologies (FRQNT).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Lavallée-Adam, M., Park, S.K.R., Martínez-Bartolomé, S. et al. From Raw Data to Biological Discoveries: A Computational Analysis Pipeline for Mass Spectrometry-Based Proteomics. J. Am. Soc. Mass Spectrom. 26, 1820–1826 (2015). https://doi.org/10.1007/s13361-015-1161-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-015-1161-7