
Abstract
The standard of care for malignant gliomas of the brain has changed very little over the last few decades, and does not offer a cure for these rare, but fatal, tumors. The field of immunotherapy has brought potent new drugs into the oncological armamentarium, and is becoming recognized as a potentially important arm in the treatment of glioblastoma for adults. Immune checkpoints are inhibitory receptors found on immune cells that, when stimulated, cause those immune cells to become quiescent. While this is a natural mechanism to prevent excessive inflammatory damage and autoimmunity in otherwise healthy tissues, cancer cells may utilize this process to grow in the absence of targeted immune destruction. Antibodies derived to block the stimulation of these negative checkpoints, allowing immune cells to remain activated and undergo effector function, are a growing area of immunotherapy. These therapies have seen much success in both the preclinical and clinical arenas for various tumors, particularly melanoma and nonsmall-cell lung cancer. Multiple clinical trials are underway to determine if these drugs have efficacy in glioblastoma. Here, we review the current evidence, from early preclinical data to lessons learned from clinical trials outside of glioblastoma, to assess the potential of immune checkpoint inhibition in the treatment of brain tumors and discuss how this therapy may be implemented with the present standard of care.
Similar content being viewed by others

Introduction
Primary brain tumors are rare and have a reported age-adjusted incidence of 22 cases per 100,000 people per year in the USA [1]. While meningioma has been the most frequently reported histology, at 36% of all primary brain tumors in patients of all ages, the broad category of gliomas are a close second, comprising 27% of all primary brain tumors. The majority of gliomas are malignant with a histologic subtype of grade IV astrocytoma, or glioblastoma (GBM). GBM is the most common malignant primary brain tumor (46%) as well as the most deadly, with a 5-year survival rate of 5%. It is important to note that brain metastases highly outnumber primary malignant brain tumors approximately 10 to 1 [2]. The most common sources of metastatic brain tumors, in descending order, are malignancies originating in the lungs (39%), breast (17%), and skin (malignant melanoma, 11%) [3]. Like GBM, prognosis following a diagnosis of metastatic brain disease is poor, with the average 2-year survival rate reported to be 8% [4].
There is currently no cure for GBM. The current standard of care (SOC) is comprised of surgical resection followed by radiation and chemotherapy. Despite these aggressive approaches, GBM always recurs, owing to its highly invasive nature. The most recent development in SOC was over a decade ago with the addition of the alkylating agent temozolomide (TMZ) as an adjuvant. Although an advancement in the treatment of GBM, adjuvant TMZ led to only a modest improvement in overall survival from 12 to 15 months [5]. Much research and effort has gone into optimizing and expanding the current SOC with novel treatment modalities.
Immune checkpoint inhibitors have gained momentum as a prominent antitumor strategy, and have generated promising results in both preclinical and clinical trials for non-central nervous system (CNS) tumors, particularly melanoma and nonsmall-cell lung cancer (NSCLC). Immune checkpoints are molecules that serve to downmodulate inflammatory reactions within the body so as to maintain immune homeostasis and prevent autoimmunity [6]. Tumors utilize immune checkpoints as major mechanisms to evade immune destruction; thus, immune checkpoint blockade serves to augment the antitumor immune response to halt or reject tumor growth and future tumor formation. The most well-characterized immune checkpoints are cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and programmed cell death 1 (PD-1), but others exist and many more yet undiscovered. Often referred to as first-generation immune checkpoint inhibitors, monoclonal antibodies that antagonize CTLA-4 or PD-1 have already reached the clinic. In early 2011, the Food and Drug Administration (FDA) approved the first immune checkpoint inhibitor, ipilimumab (a monoclonal antibody targeting CTLA-4), for the treatment of advanced melanoma [7]. Later in 2014, the FDA approved pembrolizumab and nivolumab, both anti-PD-1 monoclonal antibodies, for melanoma, NSCLC, renal cell cancer (RCC), and Hodgkin lymphoma [8, 9]. The use of these first-generation immune checkpoint inhibitors in advanced melanoma and other non-CNS tumors has produced profound survival benefits that are often long-lasting in responders.
Although malignant melanoma has seen the largest strides in terms of checkpoint inhibition, much research effort has gone into studying how these drugs can be used in treating malignant gliomas. Here, we review both preclinical and clinical evidence for the potential use of immune checkpoint blockade in glioma therapy, with a primary emphasis on anti-CTLA-4 and anti-PD-1 therapies. Then, we review the current SOC for GBM and discuss how it can potentially affect the efficacy of immune checkpoint blockade. We also discuss how potential future treatment regimens may be optimized by the use of biomarkers and combinations of different strategies in the hope of improving response rates and clinical outcomes.
Immune Checkpoints
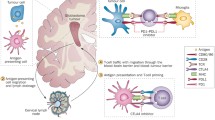
Just as immune cells may be activated by antigen and costimulatory signals, inhibitory signals also exist that act to suppress their activation and/or effector function (Fig. 1). The immune system is kept in check via these inhibitory mechanisms, referred to as immune checkpoints, in order to prevent autoimmunity and excessive destruction of healthy tissue at sites of active inflammation [6]. Cancer often utilizes these natural feedback mechanisms to avoid effective immunosurveillance, which produces a more favorable tumor microenvironment for tumor growth and formation [10]. Through direct expression of ligands of these checkpoint receptors or by stimulating their expression on other cells within the tumor microenvironment, cancer cells can effectively dampen the immune response that would otherwise benefit the host by attacking and destroying neoplastic tissue. Moreover, tumor-infiltrating lymphocytes (TILs) often co-express multiple immune checkpoint receptors due to chronic antigen stimulation within tumors, which is associated with a more severe exhausted phenotype. This T-cell exhaustion is characterized by impaired proliferative ability, effector cytokine production, and cytolytic function [11]. Thus, successful immune checkpoint blockade may require a combinatorial approach in order to generate a more robust antitumor immune response.

Immune checkpoint receptors and their cognate ligands can either inhibit or enhance the antitumor immune response. Major histocompatibility complex (MHC):antigen interaction with T cell receptor (TCR) provides either signal 1 for T-cell activation by antigen presenting cells (APCs) or subsequent recognition of tumor cells for cytolysis. Binding of costimulatory molecules B7-1 (CD80) or B7-2 (CD86) on APCs to CD28 on T cells provides signal 2 for amplification of MHC:antigen:TCR signaling. Cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) can sequester CD80/CD86 and transduce negative signals within the T cell. Programmed cell death 1 (PD-1) also inhibits T-cell effector function via distinct mechanisms when bound to its ligands (PD-L1 and PD-L2). Monoclonal antibodies targeting CTLA-4 and PD-1/PD-1/PD-L1/2 (first-generation immune checkpoint inhibitors) are currently approved for multiple tumor types and are currently being tested in clinical trials for malignant glioma. Other membrane-bound negative immune checkpoints include but are not limited to killer cell immunoglobulin-like receptors (KIRs), lymphocyte activation gene 3 (LAG-3), T-cell immunoglobulin mucin 3 (TIM-3), T-cell immunoreceptor with Ig and ITIM domains (TIGIT), and adenosine A2a receptor (A2aR). Indoleamine 2,3-dioxygenase (IDO) is a cytoplasmic protein that is the rate-limiting enzyme of tryptophan (Trp) to kynurenine (Kyn) pathway. IDO can negatively modulate T-cell responses by depleting the essential amino acid Trp and leading to the production of Kyn, which is a metabolite that can suppress T-cell function. Costimulatory receptors that enhance T-cell function upon ligand binding include inducible T-cell costimulator (ICOS), OX40 (CD134), 4-1BB, and glucocorticoid-induced TNFR family-related gene (GITR). CEACAM-1 = carcinoembryonic antigen-related cell adhesion molecule 1; PtdSer = phosphatidyl serine; PVR = poliovirus receptor (also known at CD155); ICOSL = ICOS ligand; OX40L = OX40 ligand; 4-1BBL = 4-1BB ligand; GITRL = GITR ligand; Gal-9 = galectin 9; HMG-B1 = high-mobility group protein B1
CTLA-4 and PD-1 are the most well-characterized immune checkpoints and were the first to be clinically inhibited in cancer by monoclonal antibodies (considered first-generation immune checkpoint inhibitors). However, other inhibitory immune checkpoints exist and include lymphocyte activation 3, killer cell immunoglobulin-like receptors (KIRs), T-cell immunoglobulin mucin 3 (TIM-3), T-cell immunoreceptor with Ig and ITIM domains (TIGIT), indoleamine 2,3-dioxygenase (IDO), and adenosine A2a receptor. Costimulatory receptors include inducible T-cell costimulatory, OX40 (CD134), 4-1BB, and glucocorticoid-induced TNFR family-related (GITR). The immune-mediated antitumor effects from either antagonizing inhibitory immune checkpoints or agonizing costimulatory receptors (often considered second-generation drugs) are under heavy investigation in both the preclinical and clinical settings for multiple tumor types. In the hierarchy of immune checkpoints, CTLA-4 and PD-1 appear to have an immunodominant role given the high degree of autoimmunity in knockout mice [12–16], which aligns with the severe immune-related toxicities observed in clinical trials using either anti-CTLA-4, anti-PD-1, or combined therapies (discussed later) [17]. By blocking lower-tier immune checkpoints, second-generation inhibitors are expected to have better immune side effect profiles when used as monotherapies or in combination with other immune checkpoint inhibitors.
CTLA-4
A homolog to the costimulatory molecule CD28, CTLA-4 is a glycoprotein highly yet transiently expressed on the cell surface of T cells upon activation [18, 19]. CTLA-4 is also constitutively expressed on immunosuppressive FoxP3+ regulatory T cells (Tregs) [20, 21]. CTLA-4 competitively binds to B7-1 (CD80) and B7-2 (CD86) on antigen presenting cells (APCs) with greater affinity than CD28, thus potently blocking the costimulatory signal needed to amplify T-cell receptor (TCR): major histocompatibility complex (MHC) signaling in T-cell activation [22]. Thus, while MHC–antigen complexes on APCs may bind to TCRs, secondary signals via CD28 or CTLA-4 may determine if the T cell will become activated or suppressed, respectively. Crosslinking of CTLA-4 and its ligands also transduces inhibitory signals within the T cell that further suppresses activation and effector function. CTLA-4 is known to bind to phosphatidylinositol 3-kinase (PI3K), protein tyrosine phosphatase 1, and protein phosphatase 2A via a Try–Val–Lys–Met (YVKM) motif [23]. Although not yet fully characterized, these phosphatases are postulated to propagate negative signals by counteracting TCR signaling, impairing glucose metabolism, promoting anergy, and inducing apoptosis [23]. CTLA-4 signaling is also associated with decreased interleukin (IL)-2 production, thus blunting cellular proliferation [19, 24]. Additionally, CTLA-4 has been shown to remove B7-1 (CD80) and B7-2 (CD86) on APCs through a process called transendocytosis [25]. On Tregs, CTLA-4 has an alternative function, and its stimulation causes their activation [20, 21]. Therefore, CTLA-4 suppresses the immune system from different angles by inhibiting effector T cells and activating Tregs.
Many preclinical studies have shown efficacy in targeting CTLA-4 in a variety of tumors, including melanoma, prostate cancer, and colon cancer [26–29]. In fact, blockade of CTLA-4 reversed immunosuppression in a mouse model of colon cancer, thus allowing for a robust immune response that rejected both the primary tumor, as well as subsequent tumor rechallenge [28]. The benefits of tumor regression and rebound of immune activity after blocking this receptor led to interest in using this technique for treating gliomas. Despite the relatively immune-privileged site of the CNS and the peripheral immunosuppression caused by gliomas, CTLA-4 has shown positive results in animal models of these brain tumors. Studies in a mouse model of glioma have shown that Tregs are increasingly expressed in the tumor microenvironment over time, and they have increased expression of CTLA-4 [30, 31]. After blockade of CTLA-4, there was an increase in number of CD4 T cells with improved function [32]. Furthermore, although this blockade does not suppress Treg function, it conferred CD4 T cells with resistance to Treg inhibition [32]. Significant survival benefits have been shown in mouse models when combining a CTLA-4 inhibitor with other treatments such as IL-12, tumor vaccine, and radiation therapy [33–35]. The benefits observed in these translational studies along with the successes seen in treating other non-CNS tumors in humans revealed the potential of targeting CTLA-4 in human glioma therapy.
With the advent of ipilimumab, a human monoclonal antibody specific for CTLA-4, human clinical trials targeting this immune checkpoint were made possible. Most of these studies looked at the utility of blocking CTLA-4 in malignant melanoma. In a phase III trial, Hodi et al. [7] showed that patients with previously treated metastatic melanoma had improved survival following treatment with ipilimumab, whether used in conjunction with gp100 vaccine or not. Ipilimumab also showed an improved survival benefit in conjunction with dacarbazine for patients with untreated metastatic melanoma [36]. Another trial showed a survival advantage in patients with melanoma brain metastases [37]. While ipilimumab is FDA approved for malignant melanoma, it is also being studied in clinical trials for other cancers, including malignant gliomas. There are currently two clinical trials assessing anti-CTLA-4 treatment for primary gliomas, including a phase III trial comparing the efficacy of ipilimumab and nivolumab in recurrent GBM (Table 1).
PD-1
PD-1 is expressed on B, T, and natural killer (NK) cells, and its surface expression is highly upregulated following T- and B-cell activation [38, 39]. PD-1 is also highly enriched on exhausted T cells during chronic antigen stimulation as in the setting of viral infection and cancer [11]. There are two known PD-1 ligands, PD-L1 (B7-H1 or CD274) and PD-L2 (B7-DC or CD273), and binding in the periphery suppresses effector T-cell function and mediates immune tolerance [40–42]. Upon PD-L1/2 ligation and TCR engagement, PD-1 transduces inhibitory signals via cytoplasmic domains, including an immunoreceptor tyrosine-based inhibitory motif and immunoreceoptor tyrosine-based switch motif. Although the complete story about how PD-1 signals within the cell via these motifs is unknown, there have been a couple key molecular insights. For instance, phosphorylation of the immunoreceoptor tyrosine-based switch motif has been shown to lead to the recruitment of protein tyrosine phosphatases protein tyrosine phosphatase 2 and/or protein tyrosine phosphatase 1 to attenuate TCR downstream signaling via dephosphorylation of CD3ζ, zeta-associated protein of 70 kD, and protein kinase C θ (PKC-θ) [43–46]. Moreover, CD28-mediated phosphorylation of PI3K is also reversed by PD-1 activation [47]. This serves to decrease the accumulation of antiapoptotic products (e.g., Bcl-xl) and reduce downstream protein kinase B/Akt signaling, thus impairing glucose metabolism and effector cytokine production [IL-2 and interferon (IFN)-γ] [45, 47]. PD-1 also limits T-cell proliferation by regulating gene products important for cell-cycle progression [48, 49]. Taken together, PD-1 serves to inhibit the adaptive immune response by T cells by counteracting TCR signaling, impairing T-cell expansion, lowering survival, and limiting effector function. Similar to CTLA-4, PD-1 appears to have opposite signaling effects within Tregs, where it enhances the development and maintenance of this cell population [50].
As highlighted by its role in maintaining immune homeostasis in peripheral tissue, the PD-1–PD-ligand (L)1/2 axis is a dominant mechanism hijacked by tumor cells to evade immune destruction and mediate tolerance within the tumor microenvironment [6]. PD-L1 is highly expressed on multiple tumor types, including GBM [51–54]. Myeloid cells within the tumor microenvironment also appear to be an important source of PD-L1 [55]. The prognostic value of PD-L1 expression in GBM is controversial with various studies showing mixed results [54, 56].
Mechanisms by which PD-L1 is upregulated in GBM include tumor-intrinsic oncogenic signaling and an immune negative feedback loop termed “adaptive immune resistance”. Parsa et al. [57] were the first to describe how a tumor’s genetic background could play a role in immunoresistance via enhanced PD-L1 expression. In their study, loss of functional phosphatase and tensin homolog, an important tumor suppressor gene, and abnormal PI3K/Akt signaling correlated with PD-L1 expression in GBM specimens. However, “adaptive immune resistance” refers to the mechanism by which the presence of an immune infiltrate reflexively induces PD-L1 expression in surrounding tissue (predominately through IFN-γ signaling), thus providing negative feedback on immune cells and protecting tissue from immune destruction [58, 59]. In contrast to the intrinsic tumor upregulation of PD-L1, which is always abnormal and pathologic, “adaptive immune resistance” is a normal physiologic response that is inappropriately utilized by cancer. Evidence for “adaptive immune resistance” in tumors include the induction of PD-L1 expression in tumor cells following IFN-γ treatment and the colocalization of PD-L1 expression with TILs in the tumor microenvironment [60–62].
Owing to persistent antigen stimulation in tumors, PD-1-high CD8 TILs may represent a population of impaired tumor-specific T cells that are either anergic or exhausted within the tumor microenvironment. Thus, blockade of the PD-1–PD-L1/2 interaction serves to reinvigorate this population to augment the antitumor immune response and lead to tumor rejection. In an orthotopic glioma model, anti-PD-1 therapy combined with radiation produced a long-term and durable survival advantage in mice [63]. Mice in the combined treatment arm were found to have higher levels of functional CD8 T cells within the brain than control groups. Accordingly, the improved survival advantage was lost when mice were depleted of CD8 T cells, which supports the need of these cells in mediating the antitumor response in this model. Data also suggest NK cells could also be an important immune population in PD-1 blockade, as NK cells pretreated with an anti-PD-1 antibody had increased tumor cytotoxic effects and conferred a survival advantage when injected in GBM-bearing mice compared with appropriate control arms [64].
Clinically, anti-PD-1 therapies have shown impressive responses in advanced non-CNS cancers, including melanoma, NSCLC, and RCC, with objective response rates of 31% to 40% [8, 65–67], 19% to 20% [68, 69], and 22% to 29% [70–72], respectively. Other cancers that have also demonstrated clinical benefits with anti-PD-1 therapies include Hodgkin lymphoma, and ovarian, gastric, and head and neck cancer [73]. Multiple clinical trials investigating the efficacy of anti-PD-1 and anti-PD-L1 therapies in malignant glioma are currently underway (Table 1).
Other Immune Checkpoints
Since the discovery of CTLA-4 and PD-1 and the successes seen in cancer immunotherapy through their blockade, research has been heavily focused on establishing other immune checkpoints [6]. Second-generation inhibitors that target lower-tier immune checkpoints other than CTLA-4 and PD-1, such as LAG-3, TIM-3, KIRs, IDO, and adenosine A2a receptor, are at various stages of development. As tumor immunoresistance and T-cell exhaustion are characterized by the coexpression of multiple immune checkpoint pathways, dual or multiple checkpoint blockade may generate a more robust antitumor immune response. In a glioma mouse model, blockade of TIM-3 or IDO in combination with other immune checkpoint inhibitors produced impressive response rates [74, 75]. Long-term survival was seen all mice treated with anti-TIM-3, anti-PD-1, and radiation [74]. Similar results were observed with triple therapy utilizing IDO, CTLA-4, and PD-L1 blockade [75]. These triple therapy regimens were both associated with higher levels brain-infiltrating IFN-γ+ CD8 T cells and lower Treg levels.
Additionally, just as blocking negative checkpoints is proving beneficial in boosting the antitumor immune response, there may also be utility in activating costimulatory signals. For instance, 4-1BB activation in combination with radiation and CTLA-4 blockade improved long-term survival rates in glioma-bearing mice, an effect that appeared to be dependent on CD4 T cells [35]. Using a similar model, treating mice with radiation and an agonistic antibody against GITR improved survival rates and the immune profile of brain-infiltrating T cells [76].
Toxicities Associated with Immune Checkpoint Blockade
With the expansion of clinical trials assessing the safety and efficacy of immune checkpoint inhibitors in various malignancies, drug-related side effects have become well recognized. As negative checkpoints are important natural suppressors of an immune response to prevent immunotoxicity, evidenced by CTLA-4- and PD-1-deficient mice [12–16], it is no surprise that their inhibition is associated with symptoms of immune-related dysfunction in various tissues.
With ipilimumab, dermatological manifestations, including pruritus and rash, are the most common side effects, which occur in up to 68% of patients (Fig. 2) [17, 77]. The vast majority of these symptoms are nonsevere, and they typically arise around 3 weeks after initiating treatment. Diarrhea and colitis may occur 6 weeks after the first dose in up to 46% of patients. Additionally, gastrointestinal symptoms make up the majority of severe adverse events, with severe symptoms occurring in up to 23% of patients and requiring discontinuation of therapy. Hepatotoxicity and endocrinopathies, such as hypopituitarism and hypothyroidism, both occur in < 10% of patients. Their timing following initial treatment is less consistent but typically occur after 6 to 9 weeks of therapy. While nivolumab and pembrolizumab have a similar side effect profile to ipilimumab, adverse events due to these drugs are generally less common and less severe [78]. Skin-related symptoms occur in up to 37% of patients, and severe colitis has been observed in only 1% to 2% of patients [17]. Liver and thyroid toxicities are typically similar to that of ipilimumab, but hypopituitarism may have a slightly higher incidence. Timing of initial symptoms is similar to that of ipilimumab, although they may be delayed a couple weeks. In a study by Larkin et al. [9], where patients were randomized to receiving ipilimumab and nivolumab in combination or as separate monotherapies, > 50% of the combined treatment group experienced high-grade 3 or 4 adverse events, whereas ipilimumab and nivolumab alone caused high-grade adverse events in 27% and 16% of the patients, respectively. Thus, by inhibiting multiple checkpoints, immune-related side effects appear to be more severe. Interestingly, there is evidence suggesting improved clinical outcomes in patients who experience at least grade 2 adverse events; however, these data are currently inconclusive and possibly biased by the fact that those with a clinical response to this immunotherapy remain on it for a longer period of time [79].

The majority of immune-related side effects are reversible if treated early with immunosuppressive agents and typically resolve within 6 to 8 weeks [77]. Patients who experience low-grade side effects may continue their immunotherapy regimen with the addition of medications for symptomatic relief. For example, patients may take oral antipruritic medications with topical steroids if experiencing dermatological manifestations, and those with diarrhea may be given loperamide with fluid and electrolyte repletion. If the adverse events are more severe, a 1-month oral steroid taper may be indicated. Additionally, the clinician may consider skipping a dose of the checkpoint inhibitor, decreasing the dosage, or discontinuing the treatment altogether. Although life-threatening side effects are rare, there must be a low threshold for recognizing them. In these cases, an oral steroid taper should be initiated for at least 1 month with permanent discontinuation of the immunotherapeutic agent. Thus, early recognition and intervention will be key to both alleviating these uncomfortable and potentially lethal side effects while allowing patients to continue to utilize the benefits of this anticancer therapy. Of note, endocrinopathies may not resolve with discontinuation of the immunotherapeutic agent [9, 79]. To this end, clinicians should be proactive in asking their patients if they have experienced any symptoms and should perform liver and thyroid function tests prior to each dose of treatment. As anti-CTLA-4 and anti-PD-1 therapies become increasingly investigated for the treatment of gliomas and metastatic brain tumors, their side effect profile may continue to evolve. Further insight into predicting which patients may develop these undesired symptoms and methods of preventing them will allow checkpoint inhibition to continue to grow as an important player in the treatment of brain tumors.
Biomarkers for Anti-PD-1 Response
Although immune checkpoint blockade has produced profound and objective durable tumor regressions, clinical trials in non-CNS tumors have shown that a significant proportion of patients fail to respond, or responders ultimately relapse with treatment. By examining responders versus nonresponders, clinical studies have uncovered certain biomarkers that correlate with response. Such biomarkers could have implications when using anti-PD-1 and possibly anti-CTLA-4 therapy in patients with GBM. In terms of anti-PD-1 therapy, described biomarkers include pre-existing T-cell infiltration, PD-L1 expression within the tumor, and high mutational burden [80]. It is important to note that there could be significant interaction and overlap between these biomarkers, and they may not act independently in a given patient (Fig. 3).

Potential biomarkers for clinical response to programmed cell death 1 (PD-1)/programmed cell death ligand 1 (PD-L1) blockade. From clinical trials outside of glioblastoma, CD8 T-cell infiltrate, PD-L1 expression, and high mutational burden have been described as distinct yet possibly overlapping predictors of clinical benefit to PD-1 pathway blockade. High tumor mutational burden has been associated with tumor defects in mismatch repair (MMR) proteins, mutations within the DNA polymerase epsilon gene (POLE), and environmental exposures (e.g., smoking in lung cancer and ultraviolet light in melanoma). PD-L1 can be upregulated on tumor cells via intrinsic oncogenic signaling pathways or extrinsically by infiltrating T cells through their release of interferon-γ (“adaptive immune resistance”). If a tumor lacks a pre-existing CD8 T-cell infiltrate, strategies to possibly generate one include administration of a tumor vaccine, radiation therapy, adoptive T-cell transfer, or local chemotherapy. PTEN = phosphatase and tensin homolog; PI3K = phosphatidylinositol 3-kinase; Akt = protein kinase B
Rather than allowing T cells to infiltrate the tumor, therapeutic blockade of the PD-1–PD-L1/2 axis aims to reverse or prevent the exhausted or anergic phenotype of T cells already within the tumor microenvironment. Thus, a pre-existing immune infiltrate is a prerequisite for anti-PD-1 therapy. To support this concept, Tumeh et al. [62] demonstrated that the presence and density of CD8 T cells prior to treatment predicted response to anti-PD-1 therapy in malignant melanoma. Moreover, these infiltrating T cells co-localized with PD-L1 expression at the invasive margins, which suggests engagement in the “adaptive immune resistance” mechanism and that blockade of the PD-1–PD-L1 axis could restore T-cell effector function. With regard to GBM, TILs are of variable densities and are mainly located around vasculature and tumor invasion areas within the surrounding brain parenchyma [54]. Strategies to enhance the density of TILs and possibly improve the efficacy of anti-PD-1 therapy in GBM include radiation, tumor vaccines, and adoptive T-cell therapy.
Tumor PD-L1 expression has also been shown to increase the likelihood of response to anti-PD-1 therapy. High PD-L1 expression within the tumor microenvironment may suggest the tumor heavily relies on the PD-1–PD-L1 axis for immune escape, whereas low PD-L1 levels may have alternative immune checkpoints or other immunoresistance mechanisms at play within the tumor. One of the first pivotal observations correlating tumor PD-L1 expression and anti-PD-1 response was in a phase I clinical trial by Topalian et al. [81], which investigated the safety and activity of nivolumab in multiple tumor types, including melanoma, NSCLC, RCC, colorectal cancer, and castration-resistant prostate cancer. In that study, 36% of patients with PD-L1-positive tumors had objective responses, whereas none of the patients with PD-L1 negative tumors had an objective response. Moreover, in a phase II trial looking at the efficacy of pembrolizumab in NSCLC, patients with ≥ 50% PD-L1 staining positivity had significantly higher progression-free and overall survival compared with those with < 50% PD-L1 staining positivity [68]. Thus, staining for PD-L1 could be an important selection criterion for anti-PD-1 therapy. Membranous PD-L1 expression, which is functionally more relevant than cytoplasmic expression, is found in approximately 38% and 17% of newly diagnosed and recurrent GBM, respectively [54]. The frequencies of membranous PD-L1 expression in melanoma and NSCLC are cited to be around 45% and 49%, respectively [82]. PD-L1 levels also vary depending on the GBM subtype, with the mesenchymal subtype being associated with high PD-L1 expression [54]. PD-L1 negativity is often observed in the proneural and glioma CpG island methylator phenotype subtypes [54].
Genetic alterations within tumor cells, including single amino acid substitutions, insertions, deletions, and translocations, have the propensity to become neoantigens, which are highly tumor-specific and capable of being successfully presented to the host immune system. Higher mutational burdens could be associated with a larger pool of these potential tumor neoantigens, thus giving the immune system a better opportunity to identify properly and destroy tumor cells [83]. As immune checkpoint blockade enhances the endogenous antitumor immune response, tumors with higher mutational loads are thought to be more vulnerable to immune checkpoint inhibitors. This is nicely illustrated in a clinical study with a total of 41 patients that found higher response rates to pembrolizumab in terms of observable tumor regressions and overall survival in mismatch repair (MMR)-deficient colorectal cancers, which have between 10- and 20-times more mutations than MMR-proficient tumors [84, 85]. Similarly, varying sensitivities to PD-1 blockade have been observed in melanoma and NSCLC owing to differing levels of tumor mutational burdens [86, 87]. These tumors usually carry heavy mutational loads owing to their association with environmental exposures, such as ultraviolet light for melanoma and smoking for NSCLC. Rizvi et al. [86] demonstrated that the molecular smoking signature, which is characterized by high transversion rates, was associated with a significant improvement in progression-free survival in patients with NSCLC treated with pembrolizumab. However, compared with melanoma and NSCLC, GBM has a significantly lower number of mutations per tumor (35 vs 135 and 147 for melanoma and NSCLC, respectively) [85]. Despite having a comparatively lower mutational burden, there are incidences within glioma, albeit infrequent, where mutational burden is rather high, such as loss of MMR proteins and mutations within the exonuclease proof-reading domain of the DNA polymerase epsilon gene (POLE). In a recent study, two patients with GBM with inherited defects in MMR genes displayed significant clinical responses to nivolumab [88]. Loss of MMR proteins has also been observed to occur in the setting of TMZ in low-grade glioma and GBM at disease recurrence [89–93]. TMZ-induced loss of MMR proteins is likely due to the direct genotoxic effects of the drug or drug-related epigenetic silencing. Although associated with disease progression and TMZ resistance, TMZ-induced loss of MMR proteins could possibly confer greater susceptibility to anti-PD-1 therapy or other immune checkpoint inhibitors, owing to its hypermutator phenotype. Similarly, POLE mutations, which are often associated with young age, are speculated to predict greater responses to anti-PD-1 therapy [94, 95].
Current Standard of Care
Current SOC for newly diagnosed GBM includes safe, maximal resection followed by radiation with concomitant and adjuvant TMZ [5, 96]. There is yet to be a well-established SOC for recurrent GBM. Dexamethasone is also routinely administered throughout the treatment course, especially in the postsurgical and postradiation setting, to relieve the symptoms and life-threatening complications associated with cerebral edema [97, 98]. These SOC modalities are known to interact with the immune system, and each may have an impact on the efficacy of immunotherapy in a positive or negative manner. Thus, it is paramount to determine how current SOC will influence the translation of checkpoint inhibitors to glioma or the introduction of novel glioma-specific immunotherapies.
Radiation
Radiation has been demonstrated to influence remarkably the antitumor immune response by altering the tumor microenvironment and the immunogenicity of tumor cells. In response to ionizing radiation, tumor cells upregulate surface expression of MHC class I molecules and Fas, which induces apoptosis upon interaction with its ligand [99–101]. Radiation also expands the pool of potential antigens for MHC class I loading by enhancing the degradation and production of peptides within tumor cells and generating de novo peptides [101, 102]. These changes, along with increased MHC class I expression, serve to increase the recognition and subsequent destruction of tumor cells by cytotoxic T cells. Radiation also enhances both the frequency and diversity of TCRs of TILs within the tumor microenvironment [103]. Mechanisms of heightened immune cell trafficking include radiation-induced expression of cell adhesion molecules and proinflammatory chemokines for tissue extravasation and migration, respectively [104–107].
Radiation-induced, as well as chemotherapy-induced, tumor cell death also leads to the release and expression of damage signals that activate dendritic cells (DCs). These damage signals on dying or stressed cells, along with other parameters, flag the cell death as an immunogenic, rather than tolerogenic, event [commonly referred to as immunogenic cell death (ICD)] [108, 109]. Notable damage signals include the release of the chromatin-binding high-mobility group protein B1 (HMG-B1), heat shock protein (70/90) exposure, adenosine triphosphate release, and calreticulin translocation to the cell surface. HMG-B1 is a potent adjuvant that stimulates DCs and enhances antigen processing and cross-presentation to cytotoxic T cells via Toll-like receptor 4 (TLR-4) ligation [110, 111]. HMG-B1 interaction with TLR-4 on DCs appears to be an essential component for ICD as HMG-B1 depletion or TLR-4 loss promotes tumor growth in mice after inoculation with irradiated or chemotherapy-treated (platinum-based and antracyclines) dying cancer cells [111]. TMZ has also been noted to induce ICD and synergize with DC-based vaccines in glioma mouse models [112–116].
Radiation is historically viewed as a therapy for local tumor control, but this may no longer be held true. Radiation has been observed and studied to generate a phenomenon called the abscopal effect, a systemic immune-mediated response whereby tumor regression is observed in lesions outside the radiation field [117, 118]. In GBM, the abscopal effect would be beneficial given the highly infiltrative nature of tumor cells throughout the brain parenchyma. In preclinical models, the abscopal effect has been observed in the setting of both CTLA-4 and PD-1/PD-L1 blockade, whereby combination therapy decreased the formation of distant metastases or slowed the growth of secondary nonirradiated tumors [103, 119–121]. In these models, it appears checkpoint blockade may permit the abscopal effect as radiation therapy alone failed to produce responses in secondary tumors that are of the same clonal origin as the primary tumor challenge. Additionally, the radiation schedule may influence the level of the abscopal effect. In a breast cancer mouse model, fractionated dosing but not single-dose radiation was able to generate an abscopal effect in combination with anti-CTLA-4 therapy [122].
Despite these profound local and systemic immune-stimulatory effects, radiation can also promote some immunosuppressive mechanisms. For instance, PD-L1 has been shown to be upregulated on tumor cells after radiation, which would further exhaust any T cells being trafficked to the tumor site [103, 121]. Radiation has also been shown to recruit Tregs and immunosuppressive tumor-associated macrophages within the tumor microenvironment [123–126]. Data also suggest Tregs are less sensitive to ionizing radiation, which would confer a survival advantage for this cell population in the setting of radiation therapy [127, 128]. These potential immunosuppressive mechanisms could be targeted to further augment the synergy between radiation and immunotherapy.
TMZ
Oral TMZ is commonly viewed as an immunosuppressive agent and may be antithetical to immunotherapy because it causes severe lymphopenia in patients, which would decrease the pool of tumor-specific T cells [129]. Standard TMZ and radiation produced severe drops in median CD4 counts from 664 cells/mm3 to < 300 cells/mm3 (73% of patients) and < 200 cells/mm3 (40% of patients) at 2 months post-treatment initiation in patients with high-grade gliomas [130]. The systemic depletion of CD4 T cells was also found to be long-lived as counts were persistently low at 12 months. TMZ doses that cause lymphopenia have also been shown to increase the frequency of Treg cells in glioma-bearing mice and patients with GBM [131]. On the contrary, other studies suggest that oral TMZ in low doses preferentially reduces the Treg cell compartment [114, 132, 133].
As oral TMZ is associated with severe lymphopenia, local administration may be a viable alternative that could reach adequate therapeutic levels within the tumor while limiting adverse affects [134, 135]. Intraoperative implantation of biodegradable drug-impregnated wafers of carmustine, a nitrosourea-alkylating agent, is already approved for the treatment of high-grade malignant gliomas [136]. TMZ-impregnated biodegradable wafers implanted locally into glioma-bearing rodents demonstrated lower serum TMZ levels and improved survival than oral TMZ [135]. It is important to note that all the data supporting local delivery versus systemic delivery of TMZ are from the preclinical setting with no studies in patients thus far.
Along with minimizing systemic toxicity, local delivery of TMZ has been shown to also synergize with immunotherapy in preclinical models. Intratumoral infusion of TMZ via micro-osmotic pump in conjunction with tumor vaccine in a glioma murine model demonstrated lower declines in peripheral leukocytes and generated a more favorable tumor microenvironment [137]. Compared with systemic administration of TMZ, local infusion with tumor vaccine increased the number of CD8 TILs while decreasing intratumoral myeloid-derived stem cells. Although not significant, there was also a trend of lower Treg numbers in both the blood and locally within the tumor. As local delivery leads to lower systemic levels of TMZ, this result is in line with other studies showing there is a preferential reduction of Treg with low-dose oral TMZ [114, 132, 133]. With regard to immune checkpoint blockade, local rather than systemic delivery of chemotherapy (biodegradable drug-impregnated wafers of carmustine and TMZ) synergized with PD-1 blockade by markedly improving survival rates and augmenting the antitumor immune response in glioma-bearing mice (submitted). Taken together, these findings suggest that the delivery and dosing of TMZ may be important to consider when administering this and possibly other chemotherapeutics in concert with immunotherapy regimens.
Dexamethasone
Dexamethasone, the most potent synthetic glucocorticoid, is widely used in patients with GBM because it significantly improves the morbidity and mortality associated with tumor-induced cerebral edema [97, 98]. Owing to their invasiveness and local destruction, brain tumors disrupt capillary endothelial tight junctions, which compromises blood–brain barrier integrity. Passage of fluid into the brain extracellular space leads to cerebral edema [138]. Moreover, brain tumors produce vascular endothelial growth factor and scatter factor/hepatocyte growth factor, which promote blood vessel permeability [139, 140]. Although not completely understood, dexamethasone restores the integrity of endothelial tight junctions and the blood–brain barrier [138]. Dexamethasone is stated to diminish the symptoms and improve neurologic function in approximately 75% of patients with brain metastases [141]. However, dexamethasone is a potent immunosuppressant and may modulate both the innate and adaptive immune system in a manner counterproductive to immunotherapy. For instance, glucocorticoids trigger apoptosis and maintain DCs in a more immature phenotype, which is marked by increased endocytosis, less migratory capability, poor antigen presentation, and decreased expression of both proinflammatory and costimulatory molecules [142, 143]. Along with triggering T-cell apoptosis, glucocorticoids skew T-cell polarization toward a T helper 2 over T helper 1 phenotype, which is proposed to be the dominant phenotype responsible for orchestrating the antitumor immune response. The Treg compartment is also enhanced with glucocorticoid-induced expression of IL-10, tumor growth factor-β, and intracellular FoxP3. Moreover, dexamethasone has been shown to upregulate surface expression of CTLA-4 and PD-1 in a dose-dependent manner on mouse activated T cells [144, 145]. Taken together, these glucocorticoid-driven immune effects appear to be antithetical to the goals of immunotherapy. Indeed, dexamethasone was found to decrease the frequency of TILs and abrogate the survival advantage afforded by an oncolytic viral therapy in rodent glioma models [146, 147]. To avoid these undesired effects caused by dexamethasone, other therapies could be considered to ameliorate the symptoms of cerebral edema such as antivascular endothelial growth factor therapies, including bevacizumab [148]. However, antiangiogenic therapy should strive to normalize rather than completely prune abnormal tumor vasculature in order to avoid hypoxia, which could suppress the T-cell-mediated antitumor response [149].
Future Directions
Given the poor prognosis of gliomas, we must continue to review and assess the SOC in treatment of these tumors in the hope of establishing a routine regimen with improved outcomes. An increasing understanding of the interaction between gliomas and the immune system has led to the burgeoning field of glioma immunotherapy. Reversing the immunosuppressive environment associated with these tumors is a promising arm of treatment, and future therapies may employ the use of checkpoint inhibitors in combination with radiation therapy and chemotherapy.
The addition of immune checkpoint blockade to the current SOC is an attractive strategy. Preclinical studies suggest that combination therapy is more effective than each individual therapy alone, particularly with regard to checkpoint blockade and radiation therapy. Synergism between radiation and checkpoint blockade has been shown in preclinical trials [35, 63, 74]. In addition to improved outcomes, combination therapy may allow for decreased doses of each individual treatment, thus decreasing the toxicity and side effects [150]. Interestingly, a recent phase I trial (NCT02239900) showed that combining radiation with ipilimumab for treatment of metastatic solid tumors is not only safe, but it may also promote expression of alternative immune checkpoints that may serve as additional immunotherapy targets [151]. In the same manner, the addition of checkpoint inhibition to chemotherapy has shown improved results for advanced melanoma [36]. While these synergistic combinations have been shown to be promising in preclinical studies, several clinical trials currently looking at combination therapy in a variety of cancers, including GBM, will hopefully reveal its potential as a therapeutic strategy in treating primary brain tumors in patients [152].
While many of the studies on checkpoint inhibitors assess the outcome of inhibiting a single class of these receptors, the tumor microenvironment is not static. Just as tumors are able to avoid an otherwise healthy immune system, tumor cells may adapt to overcome a specific immunotherapy. Human trials with anti-PD-1 therapy for melanoma have shown that while some individuals initially respond to therapy, many of them have recurrence and progression of the disease [66]. Proposed mechanisms of resistance to immune checkpoint blockade include but are not limited to tumor cell upregulation of alternative immune checkpoint receptors [153], secretion of immunosuppressive factors, loss of β-2 microglobulin [154, 155], and decreased sensitivity to effector cytokines such as IFN-γ [154, 156]. For instance, TIM-3 was found to be upregulated following resistance to anti-PD-1 in a lung cancer mouse model [153]. Interestingly, this TIM-3 upregulation was specifically on T cells with bound anti-PD-1 antibody, and addition of TIM-3 blockade following anti-PD-1 therapy resistance conferred a survival advantage in these mice. Thus, there may be utility in targeting multiple checkpoints instead of a single one. Preclinical studies have shown efficacy of combined therapeutic approaches, particularly with anti-PD-1 and anti-CTLA-4, in increasing tumor infiltration with immune cells, compared with individual monotherapies alone [157]. However, blocking several checkpoints has the potential to increase side effects, particularly immune-related adverse events. As previously discussed, the rate of side effects when combining ipilimumab and nivolumab may be more than double that seen in the individual therapies alone [158]. The assessment of the efficacy of combining these two immunotherapies for patients with recurrent GBM is currently underway. The combination of other immune checkpoint inhibitors and the ideal timing of using multiple therapy approaches have yet to be determined.
Finally, the variable response rates with the current checkpoint inhibitors and the growing knowledge of other immune checkpoints obviates the potential for personalized approaches to this area of immunotherapy. Biomarkers can be used to predict a patient’s response to a specific therapy and guide the clinician in the appropriate treatment regimen to prevent unnecessary costs and adverse effects [80]. The future of brain tumor therapy may involve genetic tests or tumor characterization that will determine which checkpoint blockade to utilize, and this regimen could change over time as the tumor microenvironment may adapt to one form of therapy. With continued exploration we will hopefully be able to optimize treatment for specific patients and hopefully contribute to improving outcomes in brain tumor treatment.
Conclusion
GBM is a devastating disease with a dismal prognosis. Even with current SOC, which includes safe maximal resection followed by radiation and chemotherapy, GBM always recurs and is fatal. Over the decades, much research and effort has been done in trying to prolong overall survival, but the benefits are relatively short-lived. With the recent advent and revolution of immunotherapy, especially immune checkpoint blockade, non-CNS tumors have experienced impressive responses with durable long-lasting survival benefits. Early preclinical work has demonstrated that immunotherapy may potentially hold the same promise for GBM, but more studies on the patient level are required to validate its true efficacy. With this said, clinical trials on translating immune checkpoint inhibitors and utilizing other forms of immunotherapy in GBM are currently being undertaken, and many more could be on the horizon. As GBM can develop multiple immunoresistance mechanisms, combinations using multiple checkpoint inhibitors with or without other immune-based strategies may be the most effective means in generating the most robust antitumor immune response. With this quick introduction, it will also be paramount to understand how immunotherapy regimens can be successfully incorporated into the current SOC for GBM.
References
Ostrom QT, Gittleman H, Fulop J, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2008-2012. Neuro Oncol 2015;17(Suppl. 4):iv1-iv62.
Nayak L, Lee EQ, Wen PY. Epidemiology of brain metastases. Curr Oncol Rep 2012;14:48–54.
Nussbaum ES, Djalilian HR, Cho KH, Hall WA. Brain metastases: histology, multiplicity, surgery, and survival. Cancer 1996;78:1781–1788.
Hall W, Djalilian H, Nussbaum E, Cho K, Wa H. Long-term survival with metastatic cancer to the brain. Med Ontol 2000;17:279–286.
Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352:987–996.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012;12:252–264.
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711–723.
Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015;372:2521–2532.
Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34.
Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Adv Immunol 2006;90:51–81.
Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015;15:486–499.
Waterhouse P, Penninger JM, Timms E, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 1995;270:985–988.
Chambers CA, Sullivan TJ, Allison JP. Lymphoproliferation in CTLA-4-deficient mice is mediated by costimulation-dependent activation of CD4+ T cells. Immunity 1997;7:885–895.
Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999;11:141–151.
Nishimura H, Okazaki T, Tanaka Y, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001;291:319–322.
Salama AD, Chitnis T, Imitola J, et al. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J Exp Med 2003;198:71–78.
Villadolid J, Amin A. Immune checkpoint inhibitors in clinical practice: update on management of immune-related toxicities. Transl Lung Cancer Res 2015;4:560–575.
Brunet J-F, Denizot F, Luciani M-F, et al. A new member of the immunoglobulin superfamily—CTLA-4. Nature 1987;328:267–270.
Walunas TL, Lenschow DJ, Bakker CY, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994;1:405–413.
Read S, Malmström V, Powrie F. Cytotoxic T lymphocyte–associated antigen 4 plays an essential role in the function of Cd25+Cd4+ regulatory cells that control intestinal inflammation. J Exp Med 2000;192:295-302.
Takahashi T, Tagami T, Yamazaki S, et al. Immunologic self-tolerance maintained by Cd25+Cd4+regulatory t cells constitutively expressing cytotoxic T lymphocyte–associated antigen 4. J Exp Med 2000;192.
Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1994;1:793–801.
Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev 2009;12–26.
Krummel MF, Allison JP. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J Exp Med 1996;183:2533-2540.
Qureshi OS, Zheng Y, Nakamura K, et al. Trans- endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 2011;332:600–603.
Hurwitz AA, Yu TF, Leach DR, Allison JP. CTLA-4 blockade synergizes with tumor-derived granulocyte-macrophage colony-stimulating factor for treatment of an experimental mammary carcinoma. Proc Natl Acad Sci U S A 1998;95:10067–10071.
Kwon ED, Hurwitz AA, Foster BA, et al. Manipulation of T cell costimulatory and inhibitory signals for immunotherapy of prostate cancer. Proc Natl Acad Sci U S A 1997;94:8099–8103.
Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996;271:1734–1736.
van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med 1999;190:355-366.
El Andaloussi A, Han Y, Lesniak MS. Prolongation of survival following depletion of CD4+CD25+ regulatory T cells in mice with experimental brain tumors. J Neurosurg 2006;105:430–437.
Grauer OM, Nierkens S, Bennink E, et al. CD4+FoxP3+ regulatory T cells gradually accumulate in gliomas during tumor growth and efficiently suppress antiglioma immune responsesin vivo. Int J Cancer 2007;121:95–105.
Fecci PE, Ochiai H, Mitchell DA, et al. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin Cancer Res 2007;13:2158–2167.
vom Berg J, Vrohlings M, Haller S, et al. Intratumoral IL-12 combined with CTLA-4 blockade elicits T cell–mediated glioma rejection. J Exp Med 2013;210:2803–2811.
Agarwalla P, Barnard Z, Fecci P, Dranoff G, Curry WT. Sequential immunotherapy by vaccination with GM-CSF-expressing glioma cells and CTLA-4 blockade effectively treats established murine intracranial tumors. J Immunother 2012;35:385–389.
Belcaid Z, Phallen JA, Zeng J, et al. Focal radiation therapy combined with 4-1BB activation and CTLA-4 blockade yields long-term survival and a protective antigen-specific memory response in a murine glioma model. PLOS ONE 2014;9:e101764.
Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011;364:2517–2526.
Margolin K, Ernstoff MS, Hamid O, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol 2012;13:459–465.
Agata Y, Kawasaki A, Nishimura H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol 1996;8:765–772.
Vibhakar R, Juan G, Traganos F, Darzynkiewicz Z, Finger LR. Activation-induced expression of human programmed death-1 gene in T-lymphocytes. Exp Cell Res 1997;232:25–28.
Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 2000;192:1027–1034.
Latchman Y, Wood CR, Chernova T, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol 2001;2:261–268.
Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008;26:677–704.
Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci U S A 2001;98:13866–13871.
Sheppard KA, Fitz LJ, Lee JM, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3ζ signalosome and downstream signaling to PKCθ FEBS Lett 2004;574:37–41.
Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol 2004;173:945–954.
Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med 2012;209:1201–1217.
Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 2005;25:9543–9553.
Patsoukis N, Brown J, Petkova V, Liu F, Li L, Boussiotis VA. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci Signal 2012;5:ra46.
Patsoukis N, Sari D, Boussiotis VA. PD-1 inhibits T cell proliferation by upregulating p27 and p15 and suppressing Cdc25A. Cell Cycle 2012;11:4305–4309.
Francisco LM, Salinas VH, Brown KE, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med 2009;206:3015–3029.
Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol 2008;8:467–477.
Wintterle S, Schreiner B, Mitsdoerffer M, et al. Expression of the B7-related molecule B7-H1 by glioma cells : a potential mechanism of immune paralysis. Cancer Res 2003;63:7462–7467.
Wilmotte R, Burkhardt K, Kindler V, et al. B7-homolog 1 expression by human glioma: a new mechanism of immune evasion. Neuroreport 2005;16:1081–1085.
Berghoff AS, Kiesel B, Widhalm G, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol 2015;17:1064–1075.
Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin. Cancer Res 2013;19:3165–3175.
Nduom EK, Wei J, Yaghi NK, et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol 2016;18:195–205.
Parsa AT, Waldron JS, Panner A, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med 2007;13:84–88.
Kim J, Myers AC, Chen L, et al. Constitutive and inducible expression of B7 family of ligands by human airway epithelial cells. Am J Respir Cell Mol Biol 2005;33:280–289.
Lee SK, Seo SH, Kim BS, et al. IFN-gamma regulates the expression of B7-H1 in dermal fibroblast cells. J Dermatol Sci 2005;40:95–103.
Han SJ, Ahn BJ, Waldron JS, et al. Gamma interferon-mediated superinduction of B7-H1 in PTEN-deficient glioblastoma: a paradoxical mechanism of immune evasion. Neuroreport 2009;20:1597–1602.
Taube JM, Anders RA, Young GD, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med 2012;4:127ra37.
Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–571.
Zeng J, See AP, Phallen J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys 2013;86:343–349.
Huang BY, Zhan YP, Zong WJ, et al. The PD-1/B7-H1 pathway modulates the natural killer cells versus mouse glioma stem cells. PLOS ONE 2015;10:1–14.
Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014;32:1020–1030.
Ribas A, Hamid O, Daud A, et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA 2016;315:1600.
Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320–330.
Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med 2015;372:2018–2028.
Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med 2015;373:123–135.
Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;1803–1813.
Motzer RJ, Rini BI, McDermott DF, et al. Nivolumab for metastatic renal cell carcinoma: results of a randomized phase II trial. J Clin Oncol 2015;33:1430–1437.
McDermott DF, Drake CG, Sznol M, et al. Survival, durable response, and long-term safety in patients with previously treated advanced renal cell carcinoma receiving nivolumab. J Clin Oncol 2015;33:2013–2020.
Lipson EJ, Forde PM, Hammers HJ, Emens LA, Taube JM, Topalian SL. Antagonists of PD-1 and PD-L1 in cancer treatment. Semin Oncol 2015;42:587–600.
Kim JE, Patel MA, Mangraviti A, et al. Combination therapy with anti-PD-1, anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin Cancer Res 2016;23:124-136.
Wainwright DA, Chang AL, Dey M, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res 2014;20:5290–5301.
Patel MA, Kim JE, Theodros D, et al. Agonist anti-GITR monoclonal antibody and stereotactic radiation induce immune-mediated survival advantage in murine intracranial glioma. J Immunother Cancer 2016;4:28.
Weber JS, Dummer R, de Pril V, Lebbé C, Hodi FS. Patterns of onset and resolution of immune-related adverse events of special interest with ipilimumab. Cancer 2013;119:1675–1682.
Kottschade L, Brys A, Peikert T, et al. A multidisciplinary approach to toxicity management of modern immune checkpoint inhibitors in cancer therapy. Melanoma Res 2016;26:469–480.
Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol 2012;30:2691–2697.
Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer 2016;16:275–287.
Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–2454.
Grosso J, Horak CE, Inzunza D, et al. Association of tumor PD-L1 expression and immune biomarkers with clinical activity in patients (pts) with advanced solid tumors treated with nivolumab. J Clin Oncol 2013;31:abst3016.
Mcgranahan N, Furness AJS, Rosenthal R, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016;351:1463–1469.
Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015;372:2509–2520.
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz Jr. LA, Kinzler KW. Cancer genome landscapes. Science 2013;339:1546–1558.
Rizvi NA, Hellmann MD, Snyder A, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348:124–128.
Johnson DB, Frampton GM, Rioth MJ, et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol Res 2016;959–968.
Bouffet E, Larouche V, Campbell BB, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol 2016;34:2206–2211.
van Thuijl HF, Mazor T, Johnson BE, et al. Evolution of DNA repair defects during malignant progression of low-grade gliomas after temozolomide treatment. Acta Neuropathol 2015;129:597–607.
Stark AM, Doukas A, Hugo H-H, Mehdorn HM. The expression of mismatch repair proteins MLH1, MSH2 and MSH6 correlates with the Ki67 proliferation index and survival in patients with recurrent glioblastoma. Neurol Res 2010;6412:2180.
Yip S, Miao J, Cahill DP, et al. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res 2009;15:4622–4629.
Hunter C, Smith R, Cahill DP, et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res 2006;66:3987–3991.
Cahill DP, Levine KK, Betensky RA, et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin Cancer Res 2007;13:2038–2045.
Erson-Omay EZ, Cąʇlayan AO, Schultz N, et al. Somatic POLE mutations cause an ultramutated giant cell high-grade glioma subtype with better prognosis. Neuro Oncol 2015;17:1356–1364.
Ahn S, Ahmad AA, Kim J, et al. The somatic POLE P286R mutation defines a unique subclass of colorectal cancer featuring hypermutation , representing a potential genomic biomarker for immunotherapy. Oncotarget 2016;7:68638-68649.
Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 2009;10:459–466.
Kostaras X, Cusano F, Kline GA, Roa W, Easaw J. Use of dexamethasone in patients with high-grade glioma: a clinical practice guideline. Curr Oncol 2014;21:e493-e503.
Drappatz J, Schiff D, Kesari S, Norden AD, Wen PY. Medical management of brain tumor patients. Neurol Clin 2007;25:1035–1071.
Garnett CT, Palena C, Chakarborty M, Tsang K, Schlom J, Hodge JW. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res 2004;64:7985–7994.
Chakraborty M, Abrams SI, Camphausen K, et al. Irradiation of tumor cells up-regulates Fas and enhances CTL lytic activity and CTL adoptive immunotherapy. J Immunol 2003;170:6338–6347.
Reits EA, Hodge JW, Herberts CA, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med 2006;203:1259–1271.
Sharma A, Bode B, Wenger RH, et al. γ-Radiation promotes immunological recognition of cancer cells through increased expression of cancer-testis antigens in vitro and in vivo. PLOS ONE 2011;6:e28217.
Twyman-Saint Victor C, Rech AJ, Maity A, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015;520:373–377.
Hallahan D, Kuchibhotla J, Wyble C. Cell adhesion molecules mediate radiation-induced leukocyte adhesion to the vascular endothelium. Cancer Res 1996;5150–5155.
Handschel J, Prott FJ, Sunderkötter C, Metze D, Meyer U, Joos U. Irradiation induces increase of adhesion molecules and accumulation of β2-integrin-expressing cells in humans. Int J Radiat Oncol Biol Phys 1999;45:475–481.
Quarmby S, Hunter RD, Kumar S. Irradiation induced expression of CD31, ICAM-1 and VCAM-1 in human microvascular endothelial cells. Anticancer Res 2000;20:3375–3381.
Matsumura S, Demaria S. Up-regulation of the pro-inflammatory chemokine CXCL16 is a common response of tumor cells to ionizing radiation. Radiat Res 2010;173:418–425.
Kepp O, Senovilla L, Vitale I, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014;3:e955691.
Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol 2009;9:353–363.
Rovere-Querini P, Capobianco A, Scaffidi P, et al. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep 2004;5:825–830.
Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 2007;13:1050–1059.
Park S-D, Kim C-H, Kim C-K, et al. Cross-priming by temozolomide enhances antitumor immunity of dendritic cell vaccination in murine brain tumor model. Vaccine 2007;25:3485–3491.
Kim CH, Woo SJ, Park JS, et al. Enhanced antitumour immunity by combined use of temozolomide and TAT-survivin pulsed dendritic cells in a murine glioma. Immunology 2007;122:615–622.
Kim TG, Kim CH, Park JS, et al. Immunological factors relating to the antitumor effect of temozolomide chemoimmunotherapy in a murine glioma model. Clin Vaccine Immunol 2010;17:143–153.
Candolfi M, Yagiz K, Wibowo M, et al. Temozolomide does not impair gene therapy-mediated antitumor immunity in syngeneic brain tumor models. Clin Cancer Res 2014;20:1555–1565.
Garg AD, Vandenberk L, Koks C, et al. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell–driven rejection of high-grade glioma. Sci Transl Med 2016;8:328–327.
Postow MA, Callahan MK, Barker CA, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med 2012;366:925–931.
Golden EB, Demaria S, Schiff PB, Chachoua a, Formenti SC. An abscopal response to radiation and ipilimumab in a patient with metastatic non-small cell lung cancer. Cancer Immunol Res 2013;1:365–372.
Demaria S, Kawashima N, Yang AM, et al. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res 2005;11:728-734.
Park SS, Dong H, Liu X, et al. PD-1 restrains radiotherapy-induced abscopal effect. Cancer Immunol Res 2015;3:610-619.
Deng L, Liang H, Burnette B, et al. Irradiation and anti – PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest 2014;124:687–695.
Dewan MZ, Galloway AE, Kawashima N, et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res 2009;15:5379–5388.
Wirsdörfer F, Cappuccini F, Niazman M, et al. Thorax irradiation triggers a local and systemic accumulation of immunosuppressive CD4+ FoxP3+ regulatory T cells. Radiat Oncol 2014;9:98.
Kozin S V., Kamoun WS, Huang Y, Dawson MR, Jain RK, Duda DG. Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res 2010;70:5679–5685.
Tsai CS, Chen FH, Wang CC, et al. Macrophages from irradiated tumors express higher levels of iNOS, arginase-I and COX-2, and promote tumor growth. Int J Radiat Oncol Biol Phys. 2007;68:499–507.
Chiang C-SS, Fu SY, Wang S-CC, et al. Irradiation promotes an m2 macrophage phenotype in tumor hypoxia. Front Oncol 2012;2:89.
Qu Y, Jin S, Zhang A, Zhang B, Shi X, Wang J, Zhao Y. Gamma-ray resistance of regulatory CD4+CD25+Foxp3+ T cells in mice. Radiat Res 2010;173:148-157.
Kachikwu EL, Iwamoto KS, Liao YP, et al. Radiation enhances regulatory T cell representation. Int J Radiat Oncol Biol Phys 2011;81:1128–1135.
Su YB, Sohn S, Krown SE, et al. Selective CD4+ lymphopenia in melanoma patients treated with temozolomide: a toxicity with therapeutic implications. J Clin Oncol 2004;22:610–616.
Grossman SA, Ye X, Lesser G, et al. Immunosuppression in patients with high-grade gliomas treated with radiation and temozolomide. Clin. Cancer Res 2011;17:5473–5480.
Mitchell DA, Cui X, Schmittling RJ, et al. Monoclonal antibody blockade of IL-2Rα during lymphopenia selectively depletes regulatory T cells in mice and humans. Blood 2011;3003–3012.
Banissi C, Ghiringhelli F, Chen L, Carpentier AF. Treg depletion with a low-dose metronomic temozolomide regimen in a rat glioma model. Cancer Immunol Immunother 2009;58:1627–1634.
Ridolfi L, Petrini M, Granato AM, et al. Low-dose temozolomide before dendritic-cell vaccination reduces (specifically) CD4+CD25++Foxp3+ regulatory T-cells in advanced melanoma patients. J Transl Med 2013;11:135.
Heimberger AB, Archer GE, McLendon RE, et al. Temozolomide delivered by intracerebral microinfusion is safe and efficacious against malignant gliomas in rats. Clin Cancer Res 2000;6:4148–4153.
Brem S, Tyler B, Li K, et al. Local delivery of temozolomide by biodegradable polymers is superior to oral administration in a rodent glioma model. Cancer Chemother Pharmacol 2007;60:643–650.
Brem H, Piantadosi S, Burger PC, et al. Placebo-controlled trial of safety and efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrent gliomas. Lancet 1995;345:1008–1012.
Fritzell S, Sandén E, Eberstål S, Visse E, Darabi A, Siesjö P. Intratumoral temozolomide synergizes with immunotherapy in a T cell-dependent fashion. Cancer Immunol Immunother 2013;62:1463–1474.
Papadopoulos MC, Saadoun S, Binder DK, Manley GT, Krishna S, Verkman AS. Molecular mechanisms of brain tumor edema. Neuroscience 2004;129:1011–1020.
Machein MR, Plate KH. VEGF in brain tumors. J Neurooncol 2000;50:109–120.
Lamszus K, Laterra J, Westphal M, Rosen EM. Scatter factor/hepatocyte growth factor (SF/HGF) content and function in human gliomas. Int J Dev Neurosci 1999;17:517–530.
Ryken TC, McDermott M, Robinson PD, et al. The role of steroids in the management of brain metastases: a systematic review and evidence-based clinical practice guideline. J Neurooncol 2010;96:103–114.
Franchimont D. Overview of the actions of glucocorticoids on the immune response: A good model to characterize new pathways of immunosuppression for new treatment strategies. Ann N Y Acad Sci 2004;1024:124–137.
Flammer JR, Rogatsky I. Minireview: glucocorticoids in autoimmunity: unexpected targets and mechanisms. Mol Endocrinol 2011;25:1075–1086.
Xia M, Gasser J, Feige U. Dexamethasone enhances CTLA-4 expression during T cell activation. Cell Mol Life Sci 1999;55:1649–1656.
Wei B, Wang L, Zhao X, Du C, Guo Y, Sun Z. The upregulation of programmed death 1 on peripheral blood T cells of glioma is correlated with disease progression. Tumor Biol 2014;35:2923–2929.
Badie B, Schartner JM, Paul J, Bartley BA, Vorpahl J, Preston JK. Dexamethasone-induced abolition of the inflammatory response in an experimental glioma model: a flow cytometry study. J Neurosurg 2000;93:634–639.
Kleijn A, Kloezeman J, Treffers-Westerlaken E, et al. The in vivo therapeutic efficacy of the oncolytic adenovirus Delta24-RGD is mediated by tumor-specific immunity. PLOS ONE 2014;9:e97495.
Gil-Gil MJ, Mesia C, Rey M, Bruna J. Bevacizumab for the treatment of glioblastoma. Clin Med Insights Oncol 2013;7:123–135.
McDonald PC, Chafe SC, Dedhar S. Overcoming hypoxia-mediated tumor progression: combinatorial approaches targeting ph regulation, angiogenesis and immune dysfunction. Front Cell Dev Biol 2016;4:27.
Kocak E, Lute K, Chang X, et al. Combination therapy with anti-CTL antigen-4 and anti-4-1BB antibodies enhances cancer immunity and reduces autoimmunity. Cancer Res 2006;66:7276-7284.
Tang C, Welsh JW, de Groot P, et al. Ipilimumab with stereotactic ablative radiation therapy: phase I results and immunologic correlates from peripheral T-cells. Clin. Cancer Res 2016 Sep 20 [Epub ahead of print].
D’Souza NM, Fang P, Logan J, Yang J, Jiang W, Li J. Combining radiation therapy with immune checkpoint blockade for central nervous system malignancies. Front Oncol 2016;6:212.
Koyama S, Akbay EA, Li YY, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun 2016;7:1–9.
Zaretsky JM, Garcia-Diaz A, Shin DS, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med 2016;375:819–829.
Wang AX, Schoenhals JE, Li A, et al. Suppression of type I IFN signaling in tumors mediates resistance to anti-PD-1 treatment that can be overcome by radiotherapy. Cancer Res 2016 Nov 7 [Epub ahead of print].
Gao J, Shi LZ, Zhao H, et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 2016;167:397–404.e9.
Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A 2010;107:4275–4280.
Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013;369:122–133.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Andrew S. Luksik and Russell Maxwell contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1225 kb)
Rights and permissions
About this article

Cite this article
Luksik, A.S., Maxwell, R., Garzon-Muvdi, T. et al. The Role of Immune Checkpoint Inhibition in the Treatment of Brain Tumors. Neurotherapeutics 14, 1049–1065 (2017). https://doi.org/10.1007/s13311-017-0513-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-017-0513-3