
Abstract
It is well established that post-stroke inflammation contributes to neurovascular injury, blood–brain barrier disruption, and poor functional recovery in both animal and clinical studies. However, recent studies also suggest that several leukocyte subsets, activated during the post-stroke immune response, can exhibit both pro-injury and pro-recovery phenotypes. In accordance with these findings, B lymphocytes, or B cells, play a heterogeneous role in the adaptive immune response to stroke. This review highlights what is currently understood about the various roles of B cells, with an emphasis on stroke risk factors, as well as post-stroke injury and repair. This includes an overview of B cell functions, such as antibody production, cytokine secretion, and contribution to the immune response as antigen presenting cells. Next, evidence for B cell-mediated mechanisms in stroke-related risk factors, including hypertension, diabetes, and atherosclerosis, is outlined, followed by studies that focus on B cells during endogenous protection from stroke. Subsequently, animal studies that investigate the role of B cells in post-stroke injury and repair are summarized, and the final section describes current B cell-related clinical trials for stroke, as well as other central nervous system diseases. This review reveals the complex role of B cells in stroke, with a focus on areas for potential clinical intervention for a disease that affects millions of people globally each year.
Similar content being viewed by others


Introduction
Nearly 800,000 people in the USA experience a new or recurrent stroke every year, making stroke the fifth leading cause of death and a major cause of long-term adult disability in the US [1]. Tissue plasminogen activator is currently the only Food and Drug Administration (FDA)-approved post-stroke therapeutic [2], but delivery is required within the first 3 to 6 hours of stroke onset. Therefore, identifying mechanisms that contribute to the evolution of stroke-related injury and repair—occurring over the course of days and weeks—could expand the available window for therapeutic interventions. As highlighted by this special issue, recent focus on understanding the role of the immune system following stroke has identified several mechanisms, involving both the adaptive (e.g., B and T cells) and innate (e.g., monocytes, microglia) immune systems, that could develop into efficacious therapeutic targets. This review focuses on the roles of B cells in stroke risk and post-stroke injury and repair, with an emphasis on potential clinical interventions.
A Review of B Cell Functions
B Cells as Antibody Producers
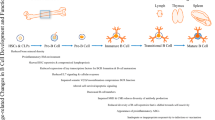
Figure 1 pictorializes the different aspects of B cell function summarized in this section. B lymphocytes (i.e., B cells) play a significant role in humoral immune responses. Naïve B cells express the primary effector molecules IgM and IgD. Upon activation, B cells switch their expression to a high-affinity, antigen-specific IgG antibody and differentiate into either memory cells or primary plasma cells (see review [3]). The “switching” to high-affinity antibodies is dependent on interactions with CD4 T cells (i.e., helper T cells), expression of CD40 on B cells, and contact with surrounding cytokines such as interferon IFN-γ or interleukin IL-4. These linked recognitions further activate both T and B cells to proliferate and clonally expand.

Summary of various B cell functions. TH = T helper; TCR = T cell receptor; MHC = major histocompatibility complex; BCR = B cell receptor; nIgM = natural IgM; MZ = marginal zone; IL = interleukin; BDNF = brain-derived neurotrophic factor; IFN = interferon; TNF = tumor necrosis factor; DC = dendritic cell; Breg = regulatory B cell; TGF = transforming growth factor; GC = Germinal center
The spleen is compartmentalized into different areas, such as follicular regions (mainly contains B cells), the marginal zone (MZ) surrounding the follicles (consists of T cells, macrophages, and B cells), the red pulp (functions as a red blood cell disposal site), and the paracortical region (consists mostly of T cells). Upon B cell activation in the spleen, there is an extrafollicular response in splenic regions, such as the red pulp and a follicular germinal center response [4, 5]. In the extrafollicular response, B cells proliferate and differentiate into short-lived, antibody-secreting plasmablasts [6]. Some of the activated B cells enter the B cell follicles and divide rapidly to form germinal centers. B cells entering germinal center reactions are known as B2 cells [7]. The germinal center provides the optimal microenvironment for various B cell processes, including proliferation, somatic hypermutation, affinity maturation, and isotype switching, all of which are essential for generating antibody diversity. B cells rearrange their DNA and alter the immunoglobulin heavy chain constant region (i.e., class-switch recombination or isotype switching) to induce different effector functions (IgA, IgG, IgM, IgD, IgE). The germinal center consists of 2 areas, a dark zone comprised of rapidly proliferating B cells, and a light zone where B cells interact with follicular dendritic cells and CD4 T cells. B cells in the light zone compete for survival signals from these dendritic cells and T cells, and are susceptible to apoptosis unless selected and expanded. This process of selective survival of B cells is termed “affinity maturation” [8]. B cells then either differentiate into plasma cells, which migrate to the bone marrow, where they continue producing higher-affinity and isotype-switched antibodies, or they differentiate into memory B cells.
Other heterogeneous populations of B cells, like B1 cells and MZ B cells, also produce antibodies to play an essential role in innate sensing of pathogens. B1 cells are generated during fetal and neonatal developmental stages and are mainly present in peritoneal and pleural cavities [9]. These innate-like B1 cells secrete broad reactivity and low-affinity natural IgM (nIgM) antibodies. In addition, MZ B cells, which are mainly present in the MZ of the spleen, also secrete nIgM [10]. These antibodies typically bind pathogens or receptors to act as their ligand (see review [11]), or activate the complement system, leading to the release of anaphylatoxins, C5a and C3a. Release of anaphylatoxins increases secretion of proinflammatory cytokines and attracts other effector cells to the site of inflammation. Polyreactive, nIgM antibodies bind and thereby coat apoptotic cells to also increase the clearance of dying or dead cells [12, 13].
B Cells as Antigen-Presenting Cells
B cells in the secondary lymphoid organs, like spleen and lymph nodes, are exposed to antigens that either enter through the afferent lymphatics, or are presented on the neighboring cell surface [14]. The B cell receptor (BCR) is an immunoglobulin on the B cell plasma membrane, which binds antigens with varying affinity, resulting in an engulfment of the antigen. The BCR works in concert with downstream signaling cascades, leading to the transcription of genes necessary for B cell function (see review [15]). The antigen, along with the BCR, is internalized in endosomal and lysosomal vesicles and the antigen is processed into peptide fragments. Accessory enzymes like proteases aid the assembly of these peptide fragments onto major histocompatibility class (MHC)-II molecules, which then traffic to the B cell’s surface to become an antigen-presenting cell (APC). The peptide fragments bound to MHC II molecules are presented to CD4 T cells for further antigen-specific T cell receptor interactions [16].
B Cells as Cytokine Producers
Apart from antibody production and antigen presentation, B cells also play a major role in modulating immune responses through cytokine secretion, as demonstrated through various animal models, especially those for autoimmune [17–19] and inflammatory [12] diseases. B regulatory cells (Bregs) suppress the immune system through the production of anti-inflammatory cytokines such as IL-10, IL-35, and transforming growth factor (TGF)-β. Bregs indirectly exert suppressive activity in a cell contact-dependent manner through the expression of inhibitory molecules, including CD40, on their surface. Bregs inhibit the expansion of inflammatory and autoreactive T cell and B cells [20–22], but require activation for cytokine production [23, 24]. Innate Bregs, derived from B1 cells, rapidly produce high amounts of IL-10 and IgM antibodies [25], while adaptive Bregs display antigen specificity and are activated by both BCR and CD40 stimulation [21]. In addition to innate and adaptive Bregs, MZ precursor B cells secrete IL-10 exclusively in response to CD40 stimulation [26].
Apart from the immunosuppressive cytokines like IL-10 and TGF-β, B cells also secrete proinflammatory cytokines, including tumor necrosis factor (TNF)-α and IFN-γ. Expression depends on the inflammatory environment or the level of anti-inflammatory cytokines such as IL-4, IL-5, and IL-13 [27]. The overall function of these cytokines is to direct Ig isotype production (i.e. IgG1 versus IgG4) [28]. Additionally, activated B cells from both humans and mice have been shown to produce neurotrophic growth factors, such as brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF), to promote the survival and differentiation of neuronal populations during central nervous system (CNS) injury [29–31].
Memory B Cells
After the initial immune response to invading pathogens, the germinal center B cells, which have undergone several rounds of positive selection (see “Tolerance Mechanisms in B Cells”), differentiate either into antibody-secreting plasma cells or memory B cells. Long-lived plasma cells home to the bone marrow, where they secrete antibodies and act as a first line of defense upon reinfection [32–34]. Memory B cells are also antigen specific, but while they express surface immunoglobulin, they do not secrete antibody at a high rate (see review [35]). Memory B cells are derived from the B cells that have undergone affinity maturation and isotype selection. Memory B cells are heterogeneous in nature, with subtypes including traditional germinal center-derived B cells, germinal center-independent memory B cells, and IgM class-switched memory B cells [36–38]. IgG+ memory B cells differentiate into plasmablasts, while IgM+ memory B cells expand during germinal center reactions [39, 40]. Chronic infection exhausts memory B cells and limits functionality [41].
Tolerance Mechanisms in B Cells
Early B cell development occurs within the bone marrow where B cells undergo both positive and negative selection, as bone marrow stromal cells provide signals to lymphoid progenitor cells through cytokine secretion and cell–cell contacts (see review [42]). These interactions prime progenitors to differentiate and commit to various lineages and start the expression of recombinase enzymes during both early and late B cell developmental stages [43]. Recombinase enzymes are involved in somatic recombination processes, which lead to either productive or non-productive B cells based on the presence of a functional heavy and light chain. B cells with unsuccessful rearrangements and improper immunoglobulin surface expression undergo apoptosis [44]. However, when B cells successfully express the cell surface receptor, signaling through the antigen receptor provides survival signals. Furthermore, multiple checkpoints during B cell development eliminate strongly self-reactive cells.
At the first checkpoint, B cell precursors expressing immunoglobulin heavy chain (IgH) are subjected to the surrogate light chain’s maintenance [45]. The surrogate chain checks how well IgH can pair with the immunoglobulin light chain (IgL) and, if paired appropriately, form the pre-B cell receptor. The light chain rearrangements, which are more complex, occur in 4 stages involving the paternal and maternal k and l loci [45, 46]. Together, B cells with a BCR comprised of light chain and heavy chain are presented with autoantigens and reach another checkpoint. Higher-affinity autoreactive B cells are deleted, while lower-affinity autoreactive B cells, particularly gut- and lung-associated B1 cells, enter the peripheral autoreactive cell pool [45]. B cells that do not recognize autoantigens home to the B cell follicles of spleen and lymph nodes to become mature B2 cells [45].
Thus, through receptor editing, B cells undergo several rounds of rearrangements, followed by positive and negative selection to ensure minimal loss of B cells while eliminating any potentially pathogenic, high-affinity, autoreactive cells. Nearly 75% of early immature B cells in humans are autoreactive, so receptor editing removes a third of these cells from the autoreactive repertoire [47]. This optimizes the chance of survival for the majority of pre-B cells, allowing them to mature into naïve B cells. However, if the initial autoreactive chains are not properly deleted during receptor editing, these B cells can revert and express the original autoreactive receptor and induce autoimmune responses [48]. With respect to autoimmune diseases, B cells are usually considered to be pathogenic owing to the secretion of autoantibodies that exacerbate tissue damage [49, 50]. Apart from secreting autoantibodies, B cells may also contribute to autoimmune disease through pro-inflammatory or anti-inflammatory cytokine production, presentation of self-antigens, regulation of other immune cells, and development of ectopic germinal centers (see reviews [51, 52]).
Downstream BCR Signaling
BCR signaling determines the selective survival of B cells based on their antigen specificity. The BCR is complexed with Igα and Igβ heterodimers [53]. When the BCR is cross-linked upon binding a receptor-specific antigen, tyrosine residues (e.g., Syk, Lyn) in the cytoplasmic BCR complex are phosphorylated by kinases to assemble adaptor proteins, kinases, guanine exchange factors, and G proteins (see reviews [54, 55]). Signal transduction processes that occur after antigen recognition by the BCR determine the magnitude of the BCR signaling. The strength of these signal transduction processes is, in turn, determined by the surface receptor density, valence, and antigen affinity of the BCR. Syk phosphorylates the Igα and Igβ complexed with the BCR, and Lyn forms a complex with the costimulatory B7 molecules and surface CD19 to reduce the activation threshold [52, 56–58]. Multiple pathways result in BCR signal transduction, including second messenger signaling and mitogen activated protein kinase pathway activation. These second messengers further activate downstream transcription factors essential for B cell function and calcium fluxes [59]. Ultimately, the B cell state and maturation stage, together with the type of antigen activating the BCR, decides the specific pathway activated downstream.
Apart from the positive signaling on antigen recognition, inhibitory receptors such as Fcγ receptors, programmed cell death 1 (CD28 homolog), and CD31 limit the intensity and duration of B cell activation through negative regulatory functions [60]. These are essential for restricting responses to foreign pathogens and discriminating against autoantigens. The disruption of these inhibitory receptors and other B cell intrinsic signaling components have been implicated in the development of autoimmune responses [61]. As discussed previously, apart from antigen recognition and binding, B cells further require secondary signals from CD4 T cells to mount a successful antigen response. MHC-II on the B cell surface binds its respective CD4 T helper cell to induce the secretion of cytokines. These cytokines trigger B cell proliferation and other downstream differentiation processes. Therefore, autoreactive B cells that escape all tolerance checkpoints and manage to enter the periphery can reach a state of unresponsiveness called anergy. Anergic B cells cannot sustain the initial antigen recognition owing to the lack of co-stimulation from CD4 T cells [47].
B Cells Contribute to the Development of Stroke Risk Factors
Clinical studies show that particular risk factors, including disease, lifestyle, sex, and age, increase the likelihood of stroke [62]. Of these risk factors—hypertension, diabetes, and atherosclerosis—are known to be potent predictors of stroke incidence, outcome, and recurrence [62]. These diseases have all been linked to changes in B cell function and phenotype, with much of the research suggesting that B cells contribute to disease development and progression [63–66] (as summarized in Fig. 2). Despite the largely deleterious effect of B cells, newer studies identify particular B cell subsets that may also be protective [65–68]. In addition to these diseases, other factors (e.g., sex, obesity, age) contribute to stroke prevalence [62], and induce both beneficial and detrimental changes to B cells [69–71], but are not specifically reviewed in this section.

Role of B cells in risk factors for stroke Ang II = angiotensin II; MS = multiple sclerosis; FOB = Follicular B cells; MZ = marginal zone; nIgM = natural IgM
Many stroke risk factors are highly comorbid [72]. For example, hypertension and diabetes are the leading risk factors for atherosclerosis, and worsen associated disease outcome [72, 73]. In line with this finding, one study showed that 57% of patients with stroke had multiple risk factors, including hypertension and diabetes, and these risk factors were also associated with increased incidence of secondary stroke [74]. The high frequency of comorbid stroke risk factors has led to the development of the “common soil” hypothesis, which suggests shared disease mechanisms for hypertension, diabetes, and atherosclerosis [75, 76], including disease-induced immune dysregulation [75]. This section will review the current research regarding B cells in hypertension, diabetes, and atherosclerosis, including B cell production of autoantibodies to disease-associated antigens as a common mechanism to these major stroke risk factors [64, 65, 77].
High Blood Pressure and Hypertension
Approximately 80 million adults in the US have hypertension (systolic blood pressure ≥ 140 mmHg) with the prevalence of this disease, and associated deaths, on the rise [62]. At the time of first stroke, approximately 77% of individuals have high blood pressure, and prehypertension is a robust predictor of future strokes [62]. For nearly 50 years, it has been known that the immune system contributes to the progression of this disease. During hypertension, there is dysregulation of the adaptive immune system, leading to global inflammation and oxidative stress in the kidneys and vasculature [78]. This inflammation has largely been attributed to a greater activation of proinflammatory T cells, suppression of regulatory T cells, and increased cytokine production [78].
Despite evidence that the immune system plays an important role in hypertension, the contributions of B cells have only recently come to light [63]. B cells isolated from patients with hypertension exhibit enhanced proliferation and activation in response to in vitro stimulation compared with normotensive individuals [79, 80]. Murine studies confirmed that immunodeficient mice that lack B cells and T cells have attenuated disease in response to angiotensin-II (Ang-II), a common rodent model of hypertension [81, 82]. B cells are also critical for the development of hypertension, as pharmacologic depletion of B cells protects against Ang-II-induced increases in systolic blood pressure, while adoptive transfer of naïve B cells restores the development of disease [63]. Additionally, B cell-deficient mice had fewer macrophages and decreased stiffening in the aorta, which is clinically an independent predictor of fatal stroke [83].
Hypertension-induced antibody production may also play a key role in pathogenesis. In hypertensive mice, there are approximately twice as many plasma cells and plasmablasts, as well as greater levels of circulating IgG and IgG deposits in the aorta, compared with wild-type (WT) mice [63]. Multiple studies corroborated that patients with hypertension have increased serum levels of IgG [84, 85], and immortalized B cells from patients have greater IgG production in vitro [79]. Patients with hypertension also present with IgG autoantibodies targeting Ang-II receptors [77, 86], with antibody titers correlated to disease severity [87]. Treatment with Ang-II receptor antagonists decreases rates of first and recurrent stroke in hypertensive patients [88], as well as reducing infarct volumes in mice [89]. These findings suggest that a further understanding of B cells in hypertension, particularly antibody production, is needed. The multiple sclerosis (MS) B cell-depleting drug, rituximab, a therapeutic antibody that targets CD20 on the B cell surface to induce apoptosis [90], has already been suggested as a therapy for patients with hypertension but has yet to be tested in the clinic [63, 91].
Diabetes Mellitus
Type 1 diabetes (T1D) is largely considered to be an incurable autoimmune condition that typically develops during childhood. It is characterized by the destruction of pancreatic insulin-secreting β cells by autoreactive T cells [64, 92]. Diabetes increases the risk of stroke regardless of age [93], and nearly triples the stroke risk in patients with a history of transient ischemic attack [94]. In addition to increasing the risk of stroke, diabetes increases stroke volume and impairs recovery [95, 96].
While T cell-mediated destruction of β cells is undoubtedly important to T1D, B cells are also critical for the development of T1D. Mice that lack B cells or receive anti-IgM therapies do not develop insulitis or diabetes [97, 98], whereas reconstitution of B cells leads to rapid expansion of pathogenic T cells [99]. Multiple methods of pharmacological depletion of B cells delay disease onset, prevent disease development, and induce long-term reversal of disease in mice (see review [90]). In new-onset patients, 4 weeks of treatment with rituximab reduced islet autoantibodies and delayed the decline of C-peptide, a protein produced during endogenous insulin secretion [100, 101]. However, this improvement was transient; by 2 years after therapy cessation, the benefits of rituximab treatment were lost [101]. It has been suggested that greater understanding of the timing and dosing of rituximab during diabetes could improve efficacy [90].
Mechanistically, B cells contribute to diabetes in several ways. MZ and FOB expand during diabetes development [102]. These subsets serve two functions. First, they differentiate into plasma cells to produce autoantibodies against insulin and other pancreatic islet antigens [103, 104]. These autoantibodies trigger a cascade of events, ultimately resulting in increased activation of cytotoxic activity of natural killer cells and CD8 T cells, which, in turn, exacerbates β cell death [103, 105]. In patients with diabetes, the presence of autoantibodies is highly predictive of T1D and often present at high levels at the time of disease onset [104, 106]. Second, MZ B cells migrate to the pancreatic lymph node where they present autoantigens to self-reactive CD4 T cells [102]. Autoreactive T cells from the serum of patients with T1D exhibit enhanced proliferation in response to islet antigens associated with diabetes pathogenesis [107–109]. Interestingly, patient with diabetes also have CNS-reactive T cells that increase the risk of developing CNS autoimmune diseases (e.g. MS [110]).
Atherosclerosis
Atherosclerosis is a condition in which lipids accumulate in medium to large arteries [111]. Intracranial atherosclerosis occurs when plaques develop in the arteries of the brain and limit blood flow throughout the brain [111]. As of 2004, approximately 70,000 ischemic strokes each year occurred owing to intracranial atherosclerosis, making it a potent risk factor for stroke [112]. Intracranial atherosclerosis is associated with high rates of recurrence, and nearly half of recurrent strokes in these patients are disabling [113]. Atherosclerosis is considered to be a chronic inflammatory disease, with lipid accumulation and adhesion of leukocytes, in particular T cells, linked to arterial inflammation [114]. T cells accumulate in vascular lesions, where they are activated by antigens [115]. In response to activation, they secrete proinflammatory cytokines that worsen inflammation and contribute to disease development [116].
Like many other risk factors for stroke, the role of B cells in atherosclerosis has, until recently, been largely overlooked. The primary B cell “players” in atherosclerosis are B1 and B2 cells, which play protective and pathogenic roles in atherosclerosis, respectively [65]. As mentioned previously, B1 cells are a small population of “innate-like” B cells that produce low-affinity natural antibodies with broad reactivity [23]. While B1 cells are a small population in mice (2–3% in spleen [117]), a similar population of B1 cells (5–10% of B cells in cord and adult blood) has been found in healthy humans [9]. The protective benefits of B1 cells are believed to be predominantly mediated through production of nIgM. In atherosclerosis, nIgM specifically targets epitopes present on damaged lipids, apoptotic cells, and necrotic cells [118]. In patients, high levels of IgM against damaged lipoproteins correlated with slower progression of atherosclerosis and low levels of nIgM against phospholipids increased the risk of stroke [119, 120]. Production of IgM appears to be necessary for protection, as adoptive transfer of IgM-deficient B1 cells into atherosclerotic mice did not reduce lesion size compared with WT B1 cell transfer [121]. In addition to IgM production, B1 cells promote clearance of apoptotic and necrotic debris in lesions [122, 123] and produce IL-10 in vivo [25].
In contrast to the protective benefits of B1 cells, B2 cells play a predominantly pathogenic role in atherosclerosis [65]. B2 cells may contribute to pathology, as depletion of B cells using a murine monoclonal anti-CD20 antibody ameliorated disease in 2 different preclinical models of atherosclerosis [124, 125]. It is interesting to note that anti-CD20 treatment selectively depletes B2 cells, while B1 cell populations are left intact [124]. Furthermore, B2-depleted mice have reduced proinflammatory cytokine production [124]. Another study showed that reconstitution of B cells in atherosclerotic mice with B cell-deficient bone marrow worsened disease [126]. While less is known about B2-mediated pathogenesis, there is some evidence that secretion of IgG autoantibodies target epitopes present on damaged lipids, such as plaque antigens, to promote inflammation. These autoantibodies are present in human atherosclerosis lesions [127], and increased levels of IgG are positively associated with disease progression in carotid atherosclerosis [128, 129]. Therefore, a partial explanation for the benefits of B cell depletion may be a reduction in pathogenic IgG autoantibodies [124], but more extensive studies are needed to determine how, if at all, IgM and/or IgG contribute to protection against atherosclerosis and disease pathology, respectively.
B Cells Also Contribute to Prestroke Neuroprotective Mechanisms
While B cells play a pathogenic role in many stroke risk factors, alterations to B cells also contribute to endogenous protection from stroke injury in animal studies [130, 131]. One method for inducing endogenous neuroprotection against stroke is preconditioning [132, 133]. Preconditioning is a prestroke intervention in which animals or patients receive brief exposure(s) to noxious stimuli that “reprogram” the way the body responds to injury [133, 134]. This reprogramming results in profound protection from stroke in animal models, including reduced infarct volumes, decreased blood–brain barrier disruption, and improved functional and neurocognitive recovery [135–137]. Many preconditioning paradigms have proven efficacious in protecting against stroke, including hypoxic preconditioning, exercise preconditioning, and ischemic preconditioning (see reviews [133, 138, 139]). Interestingly, despite the diversity of preconditioning methods, there is significant overlap in the mechanisms responsible for preconditioning-induced ischemic tolerance [132].
One way in which preconditioning protects is through modulation of the immune system (see reviews [140, 141]). Preconditioning stimuli exert stress on the body, triggering a cascade of inflammatory events that are vital for protection [141–143]. Blocking proinflammatory signaling pathways ablates the benefits of preconditioning on the brain [142–144]. Paradoxically, many of the proinflammatory pathways activated by preconditioning contribute to injury during stroke [141, 145]. Understanding how the immune system contributes to preconditioning-induced neuroprotection may further elucidate the complex interplay between the protective and pathogenic roles of the immune system in stroke. The next section will address how prototypical paradigms of preconditioning (hypoxia, exercise, and ischemia) modulate the adaptive immune system, particularly B cells.
Hypoxic Preconditioning
Many animal studies demonstrate that a single hypoxic exposure (8–11% O2) for 2–4 h induces endogenous neuroprotection (i.e., ischemic tolerance) against transient and permanent strokes [143, 146–151]. Single-exposure hypoxic preconditioning (SHP) protects against stroke by reducing infarct volumes, edema, and neurologic deficits after stroke [151]. SHP significantly modulates the immune system, altering cytokine production, chemokine expression, and overall leukocyte populations [143, 152], including an upregulation of CCL2 in cortical neurons and endothelium [143]. The chemokine CCL2 is responsible for recruitment of leukocytes to sites of inflammation in the CNS [153]. SHP upregulated CCL2 mRNA and protein at 12 h and did not induce neuroprotection in CCL2-deficient mice, revealing a necessary role for this proinflammatory chemokine [143, 154]. However, SHP also altered leukocyte populations in the blood independently of CCL2, including a significant increase in circulating B cells at 12 h after SHP, concomitant with reductions in circulating T cells, monocytes, and granulocytes [143].
While SHP induces robust protection against stroke, this protection is brief. SHP protects for a maximum of 72 h [146, 150]. However, repetitive hypoxic preconditioning (RHP) greatly extends the window of protection [155]. During RHP, mice receive hypoxic exposures varying in duration (2 or 4 h) and intensity (8% or 11% O2) for 2 weeks. RHP reduced infarct volumes by nearly half at 2 weeks after the last exposure, and, remarkably, neuroprotection persisted for 8 weeks following RHP. Despite the global effects of hypoxia, immunomodulation by RHP remained CNS-specific. RHP significantly inhibited diapedesis of CD4 T cells, monocytes, and activated macrophages into the ischemic brain, while recruiting B cells into the injured CNS [130]. Increased B cell representation may be due to an RHP-induced upregulation of vascular CXCL13, a chemokine responsible for B cell recruitment to the CNS during neuroinflammation [130, 156, 157]. A follow-up study found that RHP also upregulates cortical expression of CXCL12, a chemokine that inhibits leukocyte extravasation into the CNS [157, 158]. Blocking post-RHP CXCL12 expression with a CXCL12 receptor (CXCR4) antagonist AMD-3100 disrupted beneficial CXCL12/CCR4 signaling after RHP, and attenuated the anti-inflammatory effect of RHP in the post-stroke brain [130, 158]. While B cells remained elevated in RHP/AMD3100-treated mice, this influx did not exhibit beneficial effects on acute infarct volume. However, higher B cell representation did negatively suppress recruitment of CD4 T cells, which play a pathogenic proinflammatory role in stroke [159]. This confirms the anti-inflammatory effect of RHP-treated B cells, but raises the question of the efficacy on overall neurovascular protection against stroke. Figure 3 depicts the RHP-induced upregulation of CXCL12 at the BBB, and resulting minimizing of BBB disruption, edema, and infiltration of leukocytes in the RHP-treated brain.

Upregulation of CXCL12 at the blood–brain barrier reduces infiltration of leukocytes
While preventative treatment with hypoxia has not been tested in patients with stroke, a longitudinal study from Switzerland found that individuals who were born at higher altitudes, and thus had early exposure to hypoxic conditions, had a decreased risk of stroke [160]. Furthermore, within this population, increasing altitude correlated with decreased mortality from stroke [160]. In healthy individuals, brief repetitive exposures to hypoxia decreased circulating TNF-α for up to 1 week after final exposure, with a concomitant increase in IgG, IgM, and IgA for 4 weeks after the final exposure [161]. Although no clinical studies have specifically examined the effect of repetitive hypoxia in patients with stroke, repetitive hypoxia (alternating 1-min exposure of 9% O2 and 21% O2 for 15 min) has been used as a therapeutic in individuals with spinal cord injury. This therapy improved somatic motor function, walking speed, and walking endurance [162, 163]. Given impairments in motor function in patients with stroke, it has been suggested that repetitive hypoxia could be used as a therapeutic to improve post-stroke recovery and reduce long-term disability in this patient population [164].
Exercise Preconditioning
Individuals who exercise regularly exhibit reduced stroke risk, milder stroke injury, and greater functional recovery [165–167]. Exercise is particularly clinically relevant given that it is a noninvasive, easy way to promote protection from cerebral ischemia. Therefore, many clinical studies have investigated exercise as a preventative intervention in individuals at high risk of future strokes (see review [168]). Given the benefits of exercise in clinical populations, animal studies have sought to determine the mechanisms by which prestroke exercise protects against injury and promotes recovery (see reviews [169, 170]). In animal studies, two main methods of exercise are used for preconditioning: forced exercise (short bouts of treadmill running) and voluntary exercise (unrestricted access to running wheels). These two paradigms differentially affect stroke injury and recovery. Forced exercise results in greater reduction of infarct volume, according to a recent meta-analysis, but voluntary exercise in rats leads to greater improvements in motor recovery after stroke [137, 171].
Immunomodulation is a critical component of exercise-induced neuroprotection. Like other forms of preconditioning, ischemic tolerance after exercise preconditioning relies on activation of pro-inflammatory pathways. Over the course of 3 weeks of treadmill running, there is a progressive increase in levels of TNF-α and intracellular adhesion molecule (ICAM)-1, a protein that promotes leukocyte extravasation, in the brains of uninjured rats [172]. However, after stroke, exercised animals have decreased inflammation compared with sedentary controls. Exercise preconditioning decreased levels of endothelial ICAM-1, reduced expression of TNF-α receptors, and diminished leukocyte infiltration into the post-stroke brain [142, 173, 174]. Inhibition of inflammation with a TNF-α antibody in exercised animals reversed improvements in neurological outcome and infarct volume after stroke, supporting previous findings that TNF-α signaling plays a critical role in ischemic tolerance induced by preconditioning [142].
Acute exercise mobilizes B cells through activation of β2-adgenergic receptors by epinephrine, with greater lymphocytosis after high intensity exercise [174]. Similarly, voluntary exercise increases circulating IgM in rats, as well as enhancing B1 cell populations in the peritoneal cavity [175, 176]. In line with this finding, multiple animal studies have found that exercise improves antigen-specific antibody responses and extends the half-life of serum IgG [177–179]. Exercise also reduces apoptosis in splenic B cells isolated from mice that underwent 10 weeks of voluntary wheel running [180]. While alterations in B cell numbers can be difficult to detect in humans, there is evidence that exercise modifies B cell function, specifically increased IgM concentration following mitogen-stimulation (see review [174]). However, the benefits of exercise may be contingent on exercise intensity and duration, as sustained high-intensity exercise suppresses immune function and increases the likelihood of infection in humans [174]. This is of particular relevance as post-stroke infections worsen clinical outcomes and increase the risk of mortality for stroke patients [181].
Ischemic Preconditioning
Ischemic preconditioning is one of the oldest and most translational paradigms of preconditioning to date. Originally discovered as a way to protect against myocardial infarction in dogs, ischemic preconditioning protects against ischemic injury in other organs, including the brain (see reviews [182, 183]). Ischemic preconditioning can be achieved by 1 of 2 methods: cerebral ischemia or remote limb ischemia. Cerebral ischemic preconditioning is a method used in rodent studies in which the animal is given either a brief focal ischemic exposure or global ischemic exposure [141]. A transient occlusion of the femoral, mesenteric, or renal artery in animals, for example, induces remote ischemic preconditioning (rIPC) [169]. In clinical populations, the typical protocol for rIPC is 3 to 5 inflations of a blood pressure cuff to 200 mmHg on the forearm or leg, separated by brief periods of reperfusion [184–190]. rIPC has since been used as a safe, noninvasive method to reduce injury during coronary artery bypass surgery, abdominal aortic injury repair, pediatric cardiac surgery, and liver resection, as well as to enhance athletic performance and motor learning in healthy individuals [191–195]. Given its efficacy in decreasing ischemia–reperfusion injury, there is significant interest in application as a pre- and post-stroke intervention during ischemic stroke [196], especially in light of promising results using rIPC in other CNS diseases [187–190]. However, some clinicians express concern about our incomplete understanding of the mechanisms behind ischemic preconditioning-induced neuroprotection [190].
Our knowledge of the mechanisms of neuroprotective immunomodulation by cerebral ischemic preconditioning is limited, hindering our understanding of how B cells contribute to immune-mediated neuroprotection [141]. Ischemic preconditioning upregulates IL-1β protein, a proinflammatory cytokine that can induce B cell activation and antibody production [197, 198], and activates Toll-like receptor (TLR)-4 increases B cell proliferation, migration, and immunoglobulin secretion [199]. rIPC increases B cell populations after stroke in rats [131], mirroring our findings that B cell populations are enhanced following RHP in healthy mice. Interestingly, these findings contradict studies surrounding preconditioning that largely describe activation of acute proinflammatory signaling pathways [141], as both rIPC and RHP have, instead, an anti-inflammatory effect on the immune system that may unique to repetitive preconditioning stimuli [134].
The Role of B Cells in Stroke Injury and Recovery
Immediately after stroke there is tissue damage due to absence of blood supply to the cells. Following primary cell death, BBB integrity is compromised, with secondary reperfusion injury due to the infiltration of immune cells and the ensuing inflammatory cascade [200]. Activated endothelial cells lining the BBB express adhesion molecules to recruit peripherally circulating immune cells to bind to the endothelium and infiltrate into the brain parenchyma. This immediate and early pro-inflammatory response to stroke is mainly mediated by innate immune cells (i.e., not antigen specific) and induces cell death, tissue damage, and behavioral dysfunction [201]. This section will summarize what is known about B cell-mediated mechanisms in post-stroke injury and recovery.
CNS Immune Responses after Stroke
Infiltrating immune cells and activated tissue resident immune cells (e.g., microglia) constitute immediate, acute post-stroke inflammation [202]. In this acute phase, lymphocytes like B cells and T cells secrete cytokines and produce reactive oxygen species without regard to antigen specificity [203, 204]. Molecular signals known as danger-associated molecular pattern molecules, released by the damaged and dead neurons and other cells in the ischemic hemisphere, bind to TLRs, scavenger receptors, and other receptors on the surface of APCs for activation [205]. Dead and dying cells break down within the ischemic area within hours after stroke onset [206, 207]. Owing to BBB disruption, these cell fragments, which are normally sequestered from the peripheral circulation, act as CNS-specific antigens and elicit delayed, antigen-specific adaptive immunity after stroke [208–211]. Primed APCs present these CNS antigens to T cells and B cells in both the draining cervical lymph nodes and the spleen, and initiate the development of an adaptive immune response specific for the CNS antigens [212]. Even though by central and peripheral tolerance mechanisms most autoreactive cells have been eliminated, a few still escape into the periphery and respond to the CNS antigens [208–210, 213]. However, these post-stroke antigen-specific immune responses are largely undefined. Additional studies are required to understand if they potentiate further CNS tissue damage following stroke [208, 214–216], particularly owing to the recent identification of lymphatic vessels within the meninges [217, 218].
Post-stroke Protective Effects of B Cells
There is no clear consensus on the role of B cells in post-stroke recovery, though several animal studies propose a protective role for B cells in neuronal injury following stroke. μMT–/– mice, which have a nonsense mutation introduced into the transmembrane exon of the IgM heavy chain resulting in the total deletion of B cells [219], have larger infarct volumes, worse functional deficits, and a higher mortality rate after middle cerebral artery occlusion [220]. The ischemic hemisphere also exhibited higher numbers of activated T cells, macrophages, microglial cells, and neutrophils than WT mice. Prestroke adoptive transfer of highly enriched populations of WT B cells to μMT recipient mice was protective. Protection may be contingent on cytokine secretion, as the same neuroprotective phenotype was not observed when IL-10-deficient B cells were adoptively transferred prior to stroke [220].
Mice receiving B cells as a neurotherapeutic exhibited higher numbers of peripheral regulatory cells concomitant with lower numbers of activated pro-inflammatory T cells [221]. Regulatory T and B cells attenuate post-stroke inflammation by producing anti-inflammatory cytokines (e.g., IL-10, TGF-β). During CNS pathology, IL-10 expression in the brain increases regulatory lymphocytes and reduces CNS inflammation [51, 222–224]. LPS stimulation of B cells prior to adoptive transfer enhances IL-10 production and reduces infarct volumes, increases regulatory T cell numbers, and attenuates peripheral pro-inflammatory responses [224]. Given glatiramer acetate (i.e., copaxone) increases Breg populations in the blood and production of IL-10 [225], it has been investigated in preclinical studies of focal stroke. While Ibarra et al. found that glatiramer acetate reduced post-middle cerebral artery occlusion infarct volumes [226], this neuroprotective result has been questioned by lack of efficacy in other murine studies [227].
Post-stroke Detrimental Effects of B Cells
Other animal studies have indicated B cells as either uninvolved in, or actively injurious during post-stroke functional recovery. Mice deficient in lymphocytes [i.e., recombination activating gene (Rag1)–/–], or only deficient in CD4 T cells, CD8 T cells, B cells, or IFN-γ, were used to determine the contributions of specific lymphocyte populations to ischemia–reperfusion injury and recovery. The Rag1–/– mice had lower infarct volumes and reduced neurological deficits, but B cell-deficient mice failed to show improvements post-stroke, suggesting B cells did not significantly contribute to infarct progression [228]. Furthermore, reconstitution of RAG1−/− mice with B cells did not significantly induce post-stroke neuroprotection [229].
The occurrence of stroke increases susceptibility to dementia, in particular vascular dementia, with respect to other risk factors like diabetes, hypertension, age, and hypercholesterolemia [230–234]. However, the signaling pathways leading to post-stroke dementia development are not completely understood. Recent data report the development of cognitive deficits 7 weeks after stroke in mice [233]. In correlation with cognitive deficits, B cells aggregated in the infarct region 4 to 7 weeks post-stroke, and produced IgA and IgG antibodies. This has been verified in 4 stroke models in 2 different mouse strains. Patients with stroke also demonstrate immunoglobulin synthesis in the cerebrospinal fluid for months after stroke [235–237]. These antibodies may result in neuronal damage and further cognitive impairment by binding to the Fc receptors and activating the complement pathway, as seen in lesions of patients with MS [238]. Moreover, these antibodies could exacerbate disease by egressing into the adjacent, unaffected healthy tissue.
B cell antibody production can also directly alter neuronal function. Antibody accumulation correlated with impairments in hippocampal long-term potentiation, resulting in short-term memory deficit weeks after stroke [233]. Additionally, μMT mice did not develop delayed cognitive deficits, and rituximab attenuated cognitive decline. As opposed to the beneficial role of B cells for neuroprotection in the acute phase of stroke, these lymphocytes may play a detrimental role in post-stroke long-term cognitive impairment [233, 239]. Further studies will elucidate the dependence of CNS injury on the magnitude, location and timing of B cell subsets, in order to determine whether B cells induce neuroprotection, supporting functional plasticity, or exacerbate post-stroke recovery.
Current Stroke Clinical Trials That Potentially Impact B Cell Function
Of the clinical trials related to acute ischemic stroke, only a select few exhibit potential B cell implications in stroke. Table 1 summarizes the existing stroke clinical trials that provide reasonable sources of speculation for the role of B cells in functional recovery. To begin, several FDA-approved therapies for the treatment of MS, including fingolimod, natalizumab, and IFN-β, are now being applied to patients with acute ischemic stroke (AIS) [240]. A phase II clinical trial of natalizumab (Table 1, study 1) that assess efficacy in patients with AIS was recently completed, though the final data have not been released. However, evidence indicates that natalizumab can have a suppressive effect on B cells by inhibiting functional antibody synthesis in patients with MS [241], and thus the effect of natalizumab on B cells during AIS should be investigated. Fingolimod is now in a recruiting phase II clinical trial (Table 1, study 2) as a treatment during AIS [242]. In MS and intercerebral hemorrhage studies, fingolimod improves neurological function [243], reduces memory B cells [244], and promotes Breg phenotype and function [245]. A completed phase I clinical trial using IFN-β, with results unavailable (Table 1, study 3), investigated its safety in AIS. IFN-β knockout mice exhibit increased B cell proliferation in the blood, weight loss, and reduced strength post-stroke [246], indicating that IFN-β may attenuate inflammation in a manner related to B cell activity.
The third part of this review highlights studies that use preconditioning to minimize post-stroke injury, potentially through modulation of the adaptive immune system. Currently, several clinical trials are investigating if ischemic conditioning can decrease blood pressure (Table 1, study 4), or if postconditioning reduces infarct volume in patients with AIS (Table 1, study 5). Both studies aim to monitor serum levels of inflammatory markers that are potentially relevant to B cells, such as TNF-α, IL-6, IL-8, and IL-10, or TNF-α, ICAM-1, and glial fibrillary acidic protein, respectively. Interestingly, the interaction of LFA-1 and ICAM-1 on B cells lowers B cell activation levels [247, 248].
“Immunological Biomarkers in Patients With Acute Ischemic Stroke” is a completed clinical trial that focused on discovering immunological biomarkers in AIS in order to tailor therapy to ameliorate negative stroke outcome (Table 1, study 6). They measured circulating B cells, serum cortisol, IL-10 levels, monocyte expression of TLR4, human leukocyte antigen–antigen D-related (HLA-DR), CD86, a co-stimulatory molecule necessary for T cell activation, and very late antigen 4, an integrin that regulates the migration of leukocytes to sites of injury. When the results are published, this study will shed light on how variations in circulating B cells and their secreted cytokines can serve as biomarkers of functional recovery [249]. A clinical trial is investigating the role of serum cytokine, high motility group box 1 (Table 1, study 7). High motility group box 1 is primarily produced by mature dendritic cells to activate B cells and increase T cell reactivity, including the promotion of autoreactive B cell responses to TLR-9 ligands and DNA immune complexes by T cells [250, 251].
Associating specific white blood cell types to the pathology of AIS could ultimately allow for optimal targeted therapy, and is therefore being pursued in the CALIBER trial (Table 1, study 8). White blood cell counts can predict disease progression and outcomes of AIS [252, 253], with evidence of reduced lymphocyte circulation (B cells included) in the blood of patients following stroke [254]. Unfortunately, no clinical studies show a link between AIS, cognitive decline, and B cells, though an ongoing clinical trial (Table 1, study 9) seeks to uncover the possible relationship between inflammatory/stress compounds and post-stroke depression. It is possible that CNS-derived B cells release cytokines or antibodies that are relevant to depression [255], which will hopefully be elucidated in future clinical trials.
FDA-Approved B Cell Targeting Therapies Used in Other CNS Diseases
As described in this review, there is clear evidence in both human and mouse studies that B cells can either mediate or suppress stroke-induced pathology. By understanding the role of B cells in other CNS diseases, we may discover how B cells may truly benefit the recovering brain after stroke. In the clinic, therapies targeting B cells are used to treat CNS diseases, particularly the prototypical CNS autoimmune disease, MS. Rituximab was approved by the FDA in 1997 for follicular non-Hodgkin’s lymphoma. More recently, rituximab was found to be efficacious in patients with relapsing–remitting MS [18, 256, 257], and also reduces neurological dysfunction in the primary progressive form of MS [258].
B cell depletion successfully treats another CNS disease, anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis, which is caused by antibodies directed against the NR1 subunit of the NMDAR [259]. Patients with anti-NMDAR encephalitis may have neurological disturbances such as seizures, catatonia and coma [260, 261]. First-line treatment includes the use of cortisteroids, plasmapheresis (i.e., promotes removal of circulating autoantibodies), and intravenous administration of immunoglobulin in order to elicit an immuno-modulatory effect. A combination of rituximab and cyclophosphamide may be used as secondary treatment to deplete all circulating B cells and induces a generalized immune-suppressive effect, but initiation of clinical trials has not yet begun [261, 262]. Through studying B cell therapies for other CNS diseases, we may be able to differentiate between pathogenic and therapeutic B cells and determine how to promote the latter, resulting in effective CNS therapies in stroke.
Conclusions
The “common soil” hypothesis, first mentioned in the second section, suggests that shared disease mechanisms for hypertension, diabetes, and atherosclerosis as the greatest risk factors for stroke [75, 76]. B cell functions, in particular antibody production, are the perfect candidates to mediate these shared mechanisms. However, as outlined in this review, this commonality could also extend to protective B cell mechanisms that create an endogenous ischemic tolerance to ultimately reduce stroke injury and promote repair. Understanding how the adaptive immune system shifts from harm to benefit, and particularly which B cell functions in the at-risk or injured CNS should be therapeutically attenuated, could prove efficacious in treating a disease that affects millions globally on an annual basis.
References
Mozaffarian D, Benjamin EJ, Go AS, et al. Heart disease and stroke statistics—2015 update: a report from the American Heart Association. Circulation 2015;131:e29-322.
Kellner CP, Mocco J. Developing new stroke treatments using preclinical randomized controlled trials. World Neurosurg 2016;86:13-14.
Hoffman W, Lakkis FG, Chalasani G. B Cells, antibodies, and more. Clin J Am Soc Nephrol 2016;11:137-154.
Craft JE. Follicular helper T cells in immunity and systemic autoimmunity. Nat Rev Rheumatol 2012;8:337-347.
Weisel F, Wellmann U, Winkler TH. Autoreactive B cells get activated in extrafollicular sites. Eur J Immunol 2007;37:3330-3333.
Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol 2015 Mar;15(3):160-171.
Klein U, Dalla-Favera R. Germinal centres: role in B-cell physiology and malignancy. Nat Rev Immunol 2008 Jan;8(1):22-33.
Brink R. Germinal-center B cells in the zone. Immunity 2007 May;26(5):552-4.
Tangye SG. To B1 or not to B1: that really is still the question! Blood 2013 Jun 27;121(26):5109-5110.
Martin F, Kearney JF. Marginal-zone B cells. Nat Rev Immunol 2002 May;2(5):323-335.
Sanders J, Miguel RN, Furmaniak J, Smith BR. TSH receptor monoclonal antibodies with agonist, antagonist, and inverse agonist activities. Methods Enzymol 2010;485:393-420.
Shimomura Y, Mizoguchi E, Sugimoto K, et al. Regulatory role of B-1 B cells in chronic colitis. Int Immunol 2008 Jun;20(6):729-737.
Gronwall C, Vas J, Silverman GJ. Protective roles of natural IgM antibodies. Front Immunol 2012;3:66.
Batista FD, Harwood NE. The who, how and where of antigen presentation to B cells. Nat Rev Immunol 2009 Jan;9(1):15-27.
Wang LD, Clark MR. B-cell antigen-receptor signalling in lymphocyte development. Immunology 2003 Dec;110(4):411-420.
Chen XJ, Jensen PE. The role of B lymphocytes as antigen-presenting cells. Arch Immunol Ther Exp (Warsz) 2008 Apr;56(2):77-83.
Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol 2002 Oct;3(10):944-950.
Matsushita T, Horikawa M, Iwata Y, Tedder TF. Regulatory B cells (B10 cells) and regulatory T cells have independent roles in controlling experimental autoimmune encephalomyelitis initiation and late-phase immunopathogenesis. J Immunol 2010 Aug 15;185(4):2240-2252.
Ray A, Mann MK, Basu S, Dittel BN. A case for regulatory B cells in controlling the severity of autoimmune-mediated inflammation in experimental autoimmune encephalomyelitis and multiple sclerosis. J Neuroimmunol 2011 Jan;230(1-2):1-9.
Gray D, Gray M. What are regulatory B cells? Eur J Immunol 2010 Oct;40(10):2677-2679.
Mizoguchi A, Bhan AK. A case for regulatory B cells. J Immunol 2006 Jan 15;176(2):705-710.
Lundy SK. Killer B lymphocytes: the evidence and the potential. Inflamm Res 2009 Jul;58(7):345-357.
Zhang X. Regulatory functions of innate-like B cells. Cell Mol Immunol 2013 Mar;10(2):113-121.
Rudensky AY. Regulatory T cells and Foxp3. Immunol Rev 2011 May;241:260-268.
O'Garra A, Chang R, Go N, Hastings R, Haughton G, Howard M. Ly-1 B (B-1) cells are the main source of B cell-derived interleukin 10. Eur J Immunol 1992 Mar;22(3):711-717.
Blair PA, Chavez-Rueda KA, Evans JG, et al. Selective targeting of B cells with agonistic anti-CD40 is an efficacious strategy for the generation of induced regulatory T2-like B cells and for the suppression of lupus in MRL/lpr mice. J Immunol 2009;182(6):3492-3502.
Harris DP, Haynes L, Sayles PC, et al. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat Immunol 2000 Dec;1(6):475-482.
Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol 2008;26:261-292.
Fauchais AL, Lalloue F, Lise MC, et al. Role of endogenous brain-derived neurotrophic factor and sortilin in B cell survival. J Immunol 2008 Sep 1;181(5):3027-3038.
Edling AE, Nanavati T, Johnson JM, Tuohy VK. Human and murine lymphocyte neurotrophin expression is confined to B cells. J Neurosci Res 2004;77(5):709-717.
Kerschensteiner M, Gallmeier E, Behrens L, et al. Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: a neuroprotective role of inflammation? J Exp Med 1999;189(5):865-870.
Amanna IJ, Slifka MK. Mechanisms that determine plasma cell lifespan and the duration of humoral immunity. Immunol Rev 2010;236:125-138.
Fairfax KA, Kallies A, Nutt SL, Tarlinton DM. Plasma cell development: From B-cell subsets to long-term survival niches. Semin Immunol 2008;20(1):49-58.
Oracki SA, Walker JA, Hibbs ML, Corcoran LM, Tarlinton DM. Plasma cell development and survival. Immunol Rev 2010;237:140-159.
McHeyzer-Williams LJ, McHeyzer-Williams MG. Antigen-specific memory B cell development. Annu Rev Immunol 2005;23:487-513.
Taylor JJ, Pape KA, Jenkins MK. A germinal center-independent pathway generates unswitched memory B cells early in the primary response. J Exp Med 2012 Mar 12;209(3):597-606.
Kaji T, Ishige A, Hikida M, et al. Distinct cellular pathways select germline-encoded and somatically mutated antibodies into immunological memory. J Exp Med 2012 Oct 22;209(11):2079-2097.
Klein U, Kuppers R, Rajewsky K. Evidence for a large compartment of IgM-expressing memory B cells in humans. Blood 1997 Feb 15;89(4):1288-1298.
Dogan I, Bertocci B, Vilmont V, et al. Multiple layers of B cell memory with different effector functions. Nat Immunol 2009 Dec;10(12):1292-1299.
Pape KA, Taylor JJ, Maul RW, Gearhart PJ, Jenkins MK. Different B cell populations mediate early and late memory during an endogenous immune response. Science 2011 Mar 4;331(6021):1203-1207.
Moir S, Ho J, Malaspina A, et al. Evidence for HIV-associated B cell exhaustion in a dysfunctional memory B cell compartment in HIV-infected viremic individuals. J Exp Med 2008 Aug 4;205(8):1797-1805.
Pelanda R, Torres RM. Central B-cell tolerance: where selection begins. Cold Spring Harb Perspect Biol 2012 Apr;4(4):a007146.
Rothenberg EV. Transcriptional control of early T and B cell developmental choices. Annu Rev Immunol 2014;32:283-321.
LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood 2008 Sep 1;112(5):1570-1580.
Melchers F. Checkpoints that control B cell development. J Clin Invest 2015 Jun;125(6):2203-2210.
Vettermann C, Schlissel MS. Allelic exclusion of immunoglobulin genes: models and mechanisms. Immunol Rev 2010 Sep;237:22-42.
Cambier JC, Gauld SB, Merrell KT, Vilen BJ. B-cell anergy: from transgenic models to naturally occurring anergic B cells? Nat Rev Immunol 2007 Aug;7(8):633-643.
Luning Prak ET, Monestier M, Eisenberg RA. B cell receptor editing in tolerance and autoimmunity. Ann N Y Acad Sci 2011 Jan;1217:96-121.
Yang M, Rui K, Wang S, Lu L. Regulatory B cells in autoimmune diseases. Cell Mol Immunol 2013 Mar;10(2):122-132.
Edwards JCW, Cambridge G, Abrahams VM. Do self-perpetuating B lymphocytes drive human autoimmune disease? Immunology 1999 Jun;97(2):188-196.
Yang M, Rui K, Wang SJ, Lu LW. Regulatory B cells in autoimmune diseases. Cell Mol Immunol 2013 Mar;10(2):122-132.
Zouali M, Sarmay G. B lymphocyte signaling pathways in systemic autoimmunity - Implications for pathogenesis and treatment. Arthritis Rheum 2004 Sep;50(9):2730-2741.
Bonnerot C, Lankar D, Hanau D, et al. Role of B cell receptor Ig alpha and Ig beta subunits in MHC class II-restricted antigen presentation. Immunity 1995 Sep;3(3):335-347.
DeFranco AL. The complexity of signaling pathways activated by the BCR. Curr Opin Immunol 1997 Jun;9(3):296-308.
Kurosaki T. Regulation of BCR signaling. Mol Immunol 2011 Jun;48(11):1287-1291.
Hou P, Araujo E, Zhao T, et al. B cell antigen receptor signaling and internalization are mutually exclusive events. PLoS Biol 2006 Jul;4(7):1147-1158.
Vilen BJ, Famiglietti SJ, Carbone AM, Kay BK, Cambier JC. B cell antigen receptor desensitization—disruption of receptor coupling to tyrosine kinase activation. J Immunol 1997 Jul 1;159(1):231-243.
Cooke MP, Zeng K, Heath A, Linsley P, Howard M, Goodnow CC. B-cell tolerance is mediated by antigen receptor desensitization. J Cell Biochem 1993;??:248-???.
Niiro H, Clark EA. Regulation of B-cell fate by antigen-receptor signals. Nat Rev Immunol 2002 Dec;2(12):945-956.
Frauwirth KA, Thompson CB. Activation and inhibition of lymphocytes by costimulation. J Clin Invest 2002 Feb;109(3):295-299.
Zouali M, Sarmay G. B lymphocyte signaling pathways in systemic autoimmunity: implications for pathogenesis and treatment. Arthritis Rheum 2004 Sep;50(9):2730-2741.
Mozaffarian D, Benjamin EJ, Go AS, et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation 2016 Jan 26;133(4):e38-e360.
Chan CT, Sobey CG, Lieu M, et al. Obligatory role for B cells in the development of angiotensin II-dependent hypertension. Hypertension 2015 Nov;66(5):1023-1033.
Hinman RM, Smith MJ, Cambier JC. B cells and type 1 diabetes…in mice and men. Immunol Lett 2014 Aug;160(2):128-132.
Tsiantoulas D, Diehl CJ, Witztum JL, Binder CJ. B cells and humoral immunity in atherosclerosis. Circ Res 2014 May 23;114(11):1743-1756.
Winer DA, Winer S, Chng MH, Shen L, Engleman EG. B Lymphocytes in obesity-related adipose tissue inflammation and insulin resistance. Cell Mol Life Sci 2014 Mar;71(6):1033-1043.
Nishimura S, Manabe I, Takaki S, et al. Adipose natural regulatory B cells negatively control adipose tissue inflammation. Cell Metabolism 2013 Oct 22 [Epub ahead of print].
Harmon DB, Srikakulapu P, Kaplan JL, et al. Protective role for B-1b B cells and IgM in obesity-associated inflammation, glucose intolerance, and insulin resistance. Arterioscler Thromb Vasc Biol 2016;36:682-691.
Pennell LM, Galligan CL, Fish EN. Sex affects immunity. J Autoimmun 2012 May;38(2-3):J282-J291.
Shaikh SR, Haas KM, Beck MA, Teague H. The effects of diet-induced obesity on B cell function. Clin Exp Immunol 2015 Jan;179(1):90-99.
Rubtsova K, Rubtsov AV, Cancro MP, Marrack P. Age-associated B cells: a T-bet-dependent effector with roles in protective and pathogenic immunity. J Immunol 2015 Sep 1;195(5):1933-1937.
Sowers JR, Epstein M, Frohlich ED. Diabetes, hypertension, and cardiovascular disease: an update. Hypertension 2001 Apr;37(4):1053-1059.
Cheung BM, Li C. Diabetes and hypertension: is there a common metabolic pathway? Curr Atheroscler Rep 2012 Apr;14(2):160-166.
Lai SM, Alter M, Friday G, Sobel E. A multifactorial analysis of risk factors for recurrence of ischemic stroke. Stroke 1994 May;25(5):958-962.
Scrivo R, Vasile M, Bartosiewicz I, Valesini G. Inflammation as "common soil" of the multifactorial diseases. Autoimmun Rev 2011 May;10(7):369-374.
Tabas I, Glass CK. Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science 2013 Jan 11;339(6116):166-172.
Fu ML, Herlitz H, Schulze W, et al. Autoantibodies against the angiotensin receptor (AT1) in patients with hypertension. J Hypertens 2000 Jul;18(7):945-953.
Singh MV, Chapleau MW, Harwani SC, Abboud FM. The immune system and hypertension. Immunol Res 2014 Aug;59(1-3):243-253.
Rosskopf D, Hartung K, Hense J, Siffert W. Enhanced immunoglobulin formation of immortalized B cells from hypertensive patients. Hypertension 1995 Sep;26(3):432-435.
Siffert W, Rosskopf D, Moritz A, et al. Enhanced G protein activation in immortalized lymphoblasts from patients with essential hypertension. J Clin Invest 1995 Aug;96(2):759-766.
Guzik TJ, Hoch NE, Brown KA, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 2007 Oct 1;204(10):2449-2460.
Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P. Lymphocyte responses exacerbate angiotensin II-dependent hypertension. Am J Physiol Regul Integr Comp Physiol 2010 Apr;298(4):R1089-R1097.
Laurent S, Katsahian S, Fassot C, et al. Aortic stiffness is an independent predictor of fatal stroke in essential hypertension. Stroke 2003 May;34(5):1203-1206.
Ebringer A, Doyle AE. Raised serum IgG levels in hypertension. BMJ 1970 Apr 18;2(5702):146-48.
Hilme E, Herlitz H, Soderstrom T, Hansson L. Increased secretion of immunoglobulins in malignant hypertension. J Hypertens 1989 Feb;7(2):91-95.
Zhu F, Sun Y, Wang M, et al. Correlation between HLA-DRB1, HLA-DQB1 polymorphism and autoantibodies against angiotensin AT(1) receptors in Chinese patients with essential hypertension. Clini Cardiol 2011 May;34(5):302-308.
Siddiqui AH, Irani RA, Blackwell SC, Ramin SM, Kellems RE, Xia Y. Angiotensin receptor agonistic autoantibody is highly prevalent in preeclampsia: correlation with disease severity. Hypertension 2010 Feb;55(2):386-393.
Mogi M, Horiuchi M. Effect of angiotensin II type 2 receptor on stroke, cognitive impairment and neurodegenerative diseases. Geriatr Gerontol Int 2013 Jan;13(1):13-18.
Kobayashi T, Kawamata T, Shibata N, Okada Y, Kobayashi M, Hori T. Angiotensin II type 1 receptor blocker telmisartan reduces cerebral infarct volume and peri-infarct cytosolic phospholipase A(2) level in experimental stroke. J Neurotrauma 2009 Dec;26(12):2355-2364.
Marino E, Silveira PA, Stolp J, Grey ST. B cell-directed therapies in type 1 diabetes. Trends Immunol 2011 Jun;32(6):287-294.
Chan CT, Lieu M, Toh BH, et al. Antibodies in the pathogenesis of hypertension. BioMed Res Int 2014;2014:504045.
Roep BO, Tree TI. Immune modulation in humans: implications for type 1 diabetes mellitus. Nat Rev Endocrinol 2014 Apr;10(4):229-242.
Khoury JC, Kleindorfer D, Alwell K, et al. Diabetes mellitus: a risk factor for ischemic stroke in a large biracial population. Stroke 2013 Jun;44(6):1500-154.
Vermeer SE, Sandee W, Algra A, et al. Impaired glucose tolerance increases stroke risk in nondiabetic patients with transient ischemic attack or minor ischemic stroke. Stroke 2006 Jun;37(6):1413-1417.
Howard BV, Rodriguez BL, Bennett PH, et al. Prevention Conference VI: Diabetes and Cardiovascular disease: Writing Group I: epidemiology. Circulation. [Congresses]. 2002 May 7;105(18):e132-e137.
Toni D, De Michele M, Fiorelli M, et al. Influence of hyperglycaemia on infarct size and clinical outcome of acute ischemic stroke patients with intracranial arterial occlusion. J Neurol Sci 1994 May;123(1-2):129-133.
Serreze DV, Chapman HD, Varnum DS, et al. B lymphocytes are essential for the initiation of T cell-mediated autoimmune diabetes: analysis of a new "speed congenic" stock of NOD.Ig mu null mice. J Exp Med 1996 Nov 1;184(5):2049-2053.
Noorchashm H, Noorchashm N, Kern J, Rostami SY, Barker CF, Naji A. B-cells are required for the initiation of insulitis and sialitis in nonobese diabetic mice. Diabetes 1997 Jun;46(6):941-946.
Vong AM, Daneshjou N, Norori PY, et al. Spectratyping analysis of the islet-reactive T cell repertoire in diabetic NOD Igmu(null) mice after polyclonal B cell reconstitution. J Transl Med 2011;9:101.
Palmer JP, Fleming GA, Greenbaum CJ, et al. C-peptide is the appropriate outcome measure for type 1 diabetes clinical trials to preserve beta-cell function: report of an ADA workshop, 21-22 October 2001. Diabetes 2004 Jan;53(1):250-264.
Herold KC, Vignali DA, Cooke A, Bluestone JA. Type 1 diabetes: translating mechanistic observations into effective clinical outcomes. Nat Rev Immunol 2013 Apr;13(4):243-256.
Marino E, Batten M, Groom J, et al. Marginal-zone B-cells of nonobese diabetic mice expand with diabetes onset, invade the pancreatic lymph nodes, and present autoantigen to diabetogenic T-cells. Diabetes 2008 Feb;57(2):395-404.
Inoue Y, Kaifu T, Sugahara-Tobinai A, Nakamura A, Miyazaki J, Takai T. Activating Fc gamma receptors participate in the development of autoimmune diabetes in NOD mice. J Immunol 2007 Jul 15;179(2):764-774.
Orban T, Sosenko JM, Cuthbertson D, et al. Pancreatic islet autoantibodies as predictors of type 1 diabetes in the Diabetes Prevention Trial-Type 1. Diabetes Care 2009 Dec;32(12):2269-2274.
Harbers SO, Crocker A, Catalano G, et al. Antibody-enhanced cross-presentation of self antigen breaks T cell tolerance. J Clin Invest 2007 May;117(5):1361-1369.
Gardner SG, Gale EA, Williams AJ, et al. Progression to diabetes in relatives with islet autoantibodies. Is it inevitable? Diabetes Care 1999 Dec;22(12):2049-2054.
Abulafia-Lapid R, Elias D, Raz I, Keren-Zur Y, Atlan H, Cohen IR. T cell proliferative responses of type 1 diabetes patients and healthy individuals to human hsp60 and its peptides. J Autoimmun 1999 Mar;12(2):121-129.
Hawkes CJ, Schloot NC, Marks J, et al. T-cell lines reactive to an immunodominant epitope of the tyrosine phosphatase-like autoantigen IA-2 in type 1 diabetes. Diabetes 2000 Mar;49(3):356-366.
Congia M, Patel S, Cope AP, De Virgiliis S, Sonderstrup G. T cell epitopes of insulin defined in HLA-DR4 transgenic mice are derived from preproinsulin and proinsulin. Proc Natl Acad Sci U S A 1998 Mar 31;95(7):3833-3838.
Winer S, Astsaturov I, Cheung R, et al. Type I diabetes and multiple sclerosis patients target islet plus central nervous system autoantigens; nonimmunized nonobese diabetic mice can develop autoimmune encephalitis. J Immunol 2001 Feb 15;166(4):2831-2841.
Jeng JS, Tang SC, Liu HM. Epidemiology, diagnosis and management of intracranial atherosclerotic disease. Exp Rev Cardiovasc Ther 2010 Oct;8(10):1423-432.
Stroke O, Neuroimaging of Intracranial Atherosclerosis Trial I. Stroke outcome and neuroimaging of intracranial atherosclerosis (SONIA): design of a prospective, multicenter trial of diagnostic tests. Neuroepidemiology 2004 Jan-Apr;23(1-2):23-32.
Arenillas JF. Intracranial atherosclerosis: current concepts. Stroke 2011 Jan;42(1 Suppl.):S20-S23.
Ilhan F, Kalkanli ST. Atherosclerosis and the role of immune cells. World J Clin Cases 2015 Apr 16;3(4):345-352.
Paulsson G, Zhou X, Tornquist E, Hansson GK. Oligoclonal T cell expansions in atherosclerotic lesions of apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 2000 Jan;20(1):10-17.
Ait-Oufella H, Taleb S, Mallat Z, Tedgui A. Recent advances on the role of cytokines in atherosclerosis. Arterioscler Thromb Vasc Biol 2011 May;31(5):969-979.
Baumgarth N. The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat Rev Immunol 2011 Jan;11(1):34-46.
Kyaw T, Tipping P, Bobik A, Toh BH. Protective role of natural IgM-producing B1a cells in atherosclerosis. Trends Cardiovasc Med 2012 Feb;22(2):48-53.
Fiskesund R, Stegmayr B, Hallmans G, et al. Low levels of antibodies against phosphorylcholine predict development of stroke in a population-based study from northern Sweden. Stroke 2010 Apr;41(4):607-612.
Su J, Georgiades A, Wu R, Thulin T, de Faire U, Frostegard J. Antibodies of IgM subclass to phosphorylcholine and oxidized LDL are protective factors for atherosclerosis in patients with hypertension. Atherosclerosis 2006 Sep;188(1):160-166.
Kyaw T, Tay C, Krishnamurthi S, et al. B1a B lymphocytes are atheroprotective by secreting natural IgM that increases IgM deposits and reduces necrotic cores in atherosclerotic lesions. Circ Res 2011 Sep 30;109(8):830-840.
Ogden CA, Kowalewski R, Peng Y, Montenegro V, Elkon KB. IGM is required for efficient complement mediated phagocytosis of apoptotic cells in vivo. Autoimmunity 2005 Jun;38:259-264.
Lutz HU, Binder CJ, Kaveri S. Naturally occurring auto-antibodies in homeostasis and disease. Trends Immunol 2009 Jan;30(1):43-51.
Ait-Oufella H, Herbin O, Bouaziz JD, et al. B cell depletion reduces the development of atherosclerosis in mice. J Exp Med 2010 Aug 2;207(8):1579-1587.
Kyaw T, Tay C, Khan A, et al. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J Immunol 2010 Oct 1;185(7):4410-4419.
Major AS, Fazio S, Linton MF. B-lymphocyte deficiency increases atherosclerosis in LDL receptor-null mice. Arterioscler Thromb Vasc Biol 2002 Nov 1;22(11):1892-1898.
Yla-Herttuala S, Palinski W, Butler SW, Picard S, Steinberg D, Witztum JL. Rabbit and human atherosclerotic lesions contain IgG that recognizes epitopes of oxidized LDL. Arterioscler Thrombosis 1994 Jan;14(1):32-40.
Salonen JT, Yla-Herttuala S, Yamamoto R, et al. Autoantibody against oxidised LDL and progression of carotid atherosclerosis. Lancet 1992 Apr 11;339(8798):883-887.
Hulthe J, Bokemark L, Fagerberg B. Antibodies to oxidized LDL in relation to intima-media thickness in carotid and femoral arteries in 58-year-old subjectively clinically healthy men. Arterioscler Thromb Vasc Biol 2001 Jan;21(1):101-107.
Monson NL, Ortega SB, Ireland SJ, et al. Repetitive hypoxic preconditioning induces an immunosuppressed B cell phenotype during endogenous protection from stroke. J Neuroinflammation 2014;11:22.
Liu ZJ, Chen C, Li XR, et al. Remote ischemic preconditioning-mediated neuroprotection against stroke is associated with significant alterations in peripheral immune responses. CNS Neurosci Ther 2016 Jan;22(1):43-52.
Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci 2006 Jun;7(6):437-448.
Gidday JM. Pharmacologic preconditioning: translating the promise. Transl Stroke Res 2010;1(1):19-30.
Gidday JM. Extending injury- and disease-resistant CNS phenotypes by repetitive epigenetic conditioning. Front Neurol 2015;6:42.
Poinsatte K, Selvaraj UM, Ortega SB, et al. Quantification of neurovascular protection following repetitive hypoxic preconditioning and transient middle cerebral artery occlusion in mice. J Vis Exp 2015(99):e52675.
Hu X, Lu Y, Zhang Y, Li Y, Jiang L. Remote ischemic preconditioning improves spatial learning and memory ability after focal cerebral ischemia-reperfusion in rats. Perfusion 2013 Nov;28(6):546-551.
Egan KJ, Janssen H, Sena ES, et al. Exercise reduces infarct volume and facilitates neurobehavioral recovery: results from a systematic review and meta-analysis of exercise in experimental models of focal ischemia. Neurorehabil Neural Repair 2014 Oct;28(8):800-812.
Stetler RA, Leak RK, Gan Y, et al. Preconditioning provides neuroprotection in models of CNS disease: paradigms and clinical significance. Prog Neurobiol 2014 Mar;114:58-83.
Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol 2009 Apr;8(4):398-412.
Amantea D, Micieli G, Tassorelli C, et al. Rational modulation of the innate immune system for neuroprotection in ischemic stroke. Front Neurosci 2015;9:147.
Garcia-Bonilla L, Benakis C, Moore J, Iadecola C, Anrather J. Immune mechanisms in cerebral ischemic tolerance. Front Neurosci 2014;8:44.
Curry A, Guo M, Patel R, et al. Exercise pre-conditioning reduces brain inflammation in stroke via tumor necrosis factor-alpha, extracellular signal-regulated kinase 1/2 and matrix metalloproteinase-9 activity. Neurol Res 2010 Sep;32(7):756-762.
Stowe AM, Wacker BK, Cravens PD, et al. CCL2 upregulation triggers hypoxic preconditioning-induced protection from stroke. J Neuroinflammation 2012;9:33.
Endres M, Fan G, Hirt L, et al. Ischemic brain damage in mice after selectively modifying BDNF or NT4 gene expression. J Cereb Blood Flow Metab 2000 Jan;20(1):139-144.
Kariko K, Weissman D, Welsh FA. Inhibition of toll-like receptor and cytokine signaling--a unifying theme in ischemic tolerance. J Cereb Blood Flow Metab 2004 Nov;24(11):1288-1304.
Bernaudin M, Nedelec AS, Divoux D, MacKenzie ET, Petit E, Schumann-Bard P. Normobaric hypoxia induces tolerance to focal permanent cerebral ischemia in association with an increased expression of hypoxia-inducible factor-1 and its target genes, erythropoietin and VEGF, in the adult mouse brain. J Cereb Blood Flow Metab 2002 Apr;22(4):393-403.
Fan YY, Hu WW, Dai HB, et al. Activation of the central histaminergic system is involved in hypoxia-induced stroke tolerance in adult mice. J Cereb Blood Flow Metab 2011 Jan;31(1):305-314.
Miller BA, Perez RS, Shah AR, Gonzales ER, Park TS, Gidday JM. Cerebral protection by hypoxic preconditioning in a murine model of focal ischemia-reperfusion. Neuroreport 2001 Jun 13;12(8):1663-1669.
Tang Y, Pacary E, Freret T, et al. Effect of hypoxic preconditioning on brain genomic response before and following ischemia in the adult mouse: identification of potential neuroprotective candidates for stroke. Neurobiol Dis 2006 Jan;21(1):18-28.
Prass K, Scharff A, Ruscher K, et al. Hypoxia-induced stroke tolerance in the mouse is mediated by erythropoietin. Stroke 2003 Aug;34(8):1981-1986.
Wacker BK, Park TS, Gidday JM. Hypoxic preconditioning-induced cerebral ischemic tolerance: role of microvascular sphingosine kinase 2. Stroke 2009 Oct;40(10):3342-3348.
Liu J, Ginis I, Spatz M, Hallenbeck JM. Hypoxic preconditioning protects cultured neurons against hypoxic stress via TNF-alpha and ceramide. Am J Physiol Cell Physiol 2000 Jan;278(1):C144-C153.
Semple BD, Kossmann T, Morganti-Kossmann MC. Role of chemokines in CNS health and pathology: a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J Cereb Blood Flow Metab 2010 Mar;30(3):459-473.
Wacker BK, Perfater JL, Gidday JM. Hypoxic preconditioning induces stroke tolerance in mice via a cascading HIF, sphingosine kinase, and CCL2 signaling pathway. J Neurochem 2012 Dec;123(6):954-962.
Stowe AM, Altay T, Freie AB, Gidday JM. Repetitive hypoxia extends endogenous neurovascular protection for stroke. Ann Neurol 2011 Jun;69(6):975-985.
Huber AK, Irani DN. Targeting CXCL13 during neuroinflammation. Adv Neuroimmune Biol 2015;6(1):1-8.
Krumbholz M, Theil D, Cepok S, et al. Chemokines in multiple sclerosis: CXCL12 and CXCL13 up-regulation is differentially linked to CNS immune cell recruitment. Brain 2006 Jan;129(Pt 1):200-211.
Selvaraj UM, Ortega SB, Hu R, et al. Preconditioning-induced CXCL12 upregulation minimizes leukocyte infiltration after stroke in ischemia-tolerant mice. J Cereb Blood Flow Metab 2016 Mar 22 [Epub ahead of print].
Arumugam TV, Granger DN, Mattson MP. Stroke and T-cells. Neuromol Med 2005;7(3):229-242.
Faeh D, Gutzwiller F, Bopp M, Swiss National Cohort Study G. Lower mortality from coronary heart disease and stroke at higher altitudes in Switzerland. Circulation 2009 Aug 11;120(6):495-501.
Serebrovskaya TV, Nikolsky IS, Nikolska VV, Mallet RT, Ishchuk VA. Intermittent hypoxia mobilizes hematopoietic progenitors and augments cellular and humoral elements of innate immunity in adult men. High Alt Med Biol 2011 Fall;12(3):243-252.
Trumbower RD, Jayaraman A, Mitchell GS, Rymer WZ. Exposure to acute intermittent hypoxia augments somatic motor function in humans with incomplete spinal cord injury. Neurorehabil Neural Repair 2012 Feb;26(2):163-172.
Hayes HB, Jayaraman A, Herrmann M, Mitchell GS, Rymer WZ, Trumbower RD. Daily intermittent hypoxia enhances walking after chronic spinal cord injury: a randomized trial. Neurology 2014 Jan 14;82(2):104-113.
Dickstein R. Rehabilitation of gait speed after stroke: a critical review of intervention approaches. Neurorehabil Neural Repair 2008 Nov-Dec;22(6):649-660.
Stroud N, Mazwi TM, Case LD, et al. Prestroke physical activity and early functional status after stroke. J Neurol Neurosurg Psychiatry 2009 Sep;80(9):1019-1022.
Diep L, Kwagyan J, Kurantsin-Mills J, Weir R, Jayam-Trouth A. Association of physical activity level and stroke outcomes in men and women: a meta-analysis. J Womens Health (Larchmt) 2010 Oct;19(10):1815-1822.
Lee CD, Folsom AR, Blair SN. Physical activity and stroke risk: a meta-analysis. Stroke 2003 Oct;34(10):2475-2481.
Meschia JF, Bushnell C, Boden-Albala B, et al. Guidelines for the primary prevention of stroke: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2014 Dec;45(12):3754-3832.
Zhang F, Wu Y, Jia J. Exercise preconditioning and brain ischemic tolerance. Neuroscience 2011 Mar 17;177:170-176.
Wang X, Zhang M, Feng R, et al. Physical exercise training and neurovascular unit in ischemic stroke. Neuroscience 2014 Jun 20;271:99-107.
Ke Z, Yip SP, Li L, Zheng XX, Tong KY. The effects of voluntary, involuntary, and forced exercises on brain-derived neurotrophic factor and motor function recovery: a rat brain ischemia model. PLoS One 2011;6(2):e16643.
Ding YH, Young CN, Luan X, et al. Exercise preconditioning ameliorates inflammatory injury in ischemic rats during reperfusion. Acta Neuropathol 2005 Mar;109(3):237-246.
Ding YH, Mrizek M, Lai Q, et al. Exercise preconditioning reduces brain damage and inhibits TNF-alpha receptor expression after hypoxia/reoxygenation: an in vivo and in vitro study. Curr Neurovasc Res 2006 Nov;3(4):263-271.
Walsh NP, Gleeson M, Shephard RJ, et al. Position statement. Part one: Immune function and exercise. Exerc Immunol Rev 2011;17:6-63.
Elphick GF, Greenwood BN, Campisi J, Fleshner M. Increased serum nIgM in voluntarily physically active rats: a potential role for B-1 cells. J Appl Physiol (1985) 2003 Feb;94(2):660-667.
Elphick GF, Wieseler-Frank J, Greenwood BN, Campisi J, Fleshner M. B-1 cell (CD5+/CD11b+) numbers and nIgM levels are elevated in physically active vs. sedentary rats. J Appl Physiol 2003 Jul;95(1):199-206.
Suzuki K, Tagami K. Voluntary wheel-running exercise enhances antigen-specific antibody-producing splenic B cell response and prolongs IgG half-life in the blood. Eur J Appl Physiol 2005 Aug;94(5-6):514-519.
Liu YG, Wang SY. The enhancing effect of exercise on the production of antibody to Salmonella typhi in mice. Immunol Lett 1987 Jan;14(2):117-120.
Douglass JH. The effects of physical tracing on the immunological response in mice. J Sports Med Phys Fitness 1974 Mar;14(1):48-54.
Avula CP, Muthukumar AR, Zaman K, McCarter R, Fernandes G. Inhibitory effects of voluntary wheel exercise on apoptosis in splenic lymphocyte subsets of C57BL/6 mice. J Appl Physiol 2001;91(6):2546-2552.
Shim R, Wong CH. Ischemia, Immunosuppression and infection-tackling the predicaments of post-stroke complications. Int J Mol Sci 2016;17(1).
Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 1986 Nov;74(5):1124-1136.
Tapuria N, Kumar Y, Habib MM, Abu Amara M, Seifalian AM, Davidson BR. Remote ischemic preconditioning: a novel protective method from ischemia reperfusion injury—a review. J Surg Res 2008 Dec;150(2):304-330.
Konstantinov IE, Arab S, Kharbanda RK, et al. The remote ischemic preconditioning stimulus modifies inflammatory gene expression in humans. Physiol Genomics 2004 Sep 16;19(1):143-150.
Kharbanda RK, Peters M, Walton B, et al. Ischemic preconditioning prevents endothelial injury and systemic neutrophil activation during ischemia-reperfusion in humans in vivo. Circulation 2001 Mar 27;103(12):1624-1630.
Shimizu M, Saxena P, Konstantinov IE, et al. Remote ischemic preconditioning decreases adhesion and selectively modifies functional responses of human neutrophils. J Surg Res 2010 Jan;158(1):155-161.
Hougaard KD, Hjort N, Zeidler D, et al. Remote ischemic perconditioning as an adjunct therapy to thrombolysis in patients with acute ischemic stroke: a randomized trial. Stroke 2014 Jan;45(1):159-167.
Tulu S, Mulino M, Pinggera D, et al. Remote ischemic preconditioning in the prevention of ischemic brain damage during intracranial aneurysm treatment (RIPAT): study protocol for a randomized controlled trial. Trials 2015;16(1):594.
Meng R, Asmaro K, Meng L, et al. Upper limb ischemic preconditioning prevents recurrent stroke in intracranial arterial stenosis. Neurology 2012 Oct 30;79(18):1853-1861.
Gonzalez NR, Connolly M, Dusick JR, Bhakta H, Vespa P. Phase I clinical trial for the feasibility and safety of remote ischemic conditioning for aneurysmal subarachnoid hemorrhage. Neurosurgery 2014 Nov;75(5):590-598.
Kloner RA. Clinical application of remote ischemic preconditioning. Circulation 2009 Feb 17;119(6):776-778.
Koneru B, Shareef A, Dikdan G, et al. The ischemic preconditioning paradox in deceased donor liver transplantation-evidence from a prospective randomized single blind clinical trial. Am J Transplant 2007 Dec;7(12):2788-2796.
Jean-St-Michel E, Manlhiot C, Li J, et al. Remote preconditioning improves maximal performance in highly trained athletes. Med Sci Sports Exerc 2011 Jul;43(7):1280-1286.
Cherry-Allen KM, Gidday JM, Lee JM, Hershey T, Lang CE. Remote limb ischemic conditioning enhances motor learning in healthy humans. J Neurophysiol 2015 Jun 1;113(10):3708-3719.
Cruz RS, de Aguiar RA, Turnes T, Pereira KL, Caputo F. Effects of ischemic preconditioning on maximal constant-load cycling performance. J Appl Physiol 2015 Nov 1;119(9):961-967.
Zhao H. Hurdles to clear before clinical translation of ischemic postconditioning against stroke. Transl Stroke Res 2013 Feb;4(1):63-70.
Shin JA, Park EM, Choi JS, et al. Ischemic preconditioning-induced neuroprotection is associated with differential expression of IL-1beta and IL-1 receptor antagonist in the ischemic cortex. J Neuroimmunol 2009 Dec 10;217(1-2):14-19.
Li X, Wang X. Application of real-time polymerase chain reaction for the quantitation of interleukin-1beta mRNA upregulation in brain ischemic tolerance. Brain Res Brain Res Protoc 2000 Apr;5(2):211-217.
Bekeredjian-Ding I, Jego G. Toll-like receptors—sentries in the B-cell response. Immunology 2009 Nov;128(3):311-323.
Schroeter M, Jander S, Witte OW, Stoll G. Local immune-responses in the rat cerebral-cortex after middle cerebral-artery occlusion. J Neuroimmunol 1994 Dec;55(2):195-203.
del Zoppo GJ, Becker KJ, Hallenbeck JM. Inflammation after stroke – Is it harmful? Arch Neurol 2001 Apr;58(4):669-672.
Aktas O, Ullrich O, Infante-Duarte C, Nitsch R, Zipp F. Neuronal damage in brain inflammation. Arch Neurol 2007 Feb;64(2):185-189.
Jin R, Yang GJ, Li GH. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol 2010 May;87(5):779-789.
Kim JY, Kawabori M, Yenari MA. Innate inflammatory responses in stroke: mechanisms and potential therapeutic targets. Curr Med Chem 2014 Jun;21(18):2076-2097.
Liesz A, Dalpke A, Mracsko E, et al. DAMP signaling is a key pathway inducing immune modulation after brain injury. J Neurosci 2015 Jan 14;35(2):583-598.
Planas AM, Gomez-Choco M, Urra X, Gorina R, Caballero M, Chamorro A. Brain-derived antigens in lymphoid tissue of patients with acute stroke. J Immunol 2012 Mar 1;188(5):2156-2163.
Urra X, Miro F, Chamorro A, Planas AM. Antigen-specific immune reactions to ischemic stroke. Front Cell Neurosci 2014;8:278.
Ortega SB, Noorbhai I, Poinsatte K, et al. Stroke induces a rapid adaptive autoimmune response to novel neuronal antigens. Discov Med 2015;19(106):381-392.
Becker KJ, Kindrick DL, Lester MP, Shea C, Ye ZC. Sensitization to brain antigens after stroke is augmented by lipopolysaccharide. J Cereb Blood Flow Metab 2005 Dec;25(12):1634-1644.
Zierath D, Thullbery M, Hadwin J, et al. CNS immune responses following experimental stroke. Neurocrit Care 2010;12(2):274-284.
Becker KJ, Kalil AJ, Tanzi P, et al. Autoimmune responses to the brain after stroke are associated with worse outcome. Stroke 2011 Oct;42(10):2763-2769.
Ortega SB, Noorbhai I, Poinsatte K, et al. Stroke induces a rapid adaptive autoimmune response to novel neuronal antigens. Discov Med 2015 May;19(106):381-392.
Gee JM, Kalil A, Thullbery M, Becker KJ. Induction of immunologic tolerance to myelin basic protein prevents central nervous system autoimmunity and improves outcome after stroke. Stroke 2008 May;39(5):1575-1582.
Hallenbeck J. Adaptive immunity – Introduction. Stroke 2007 Feb;38(2):768-769.
Schwartz M, Kipnis J. Autoimmunity on alert: naturally occurring regulatory CD4(+)CD25(+) T cells as part of the evolutionary compromise between a 'need' and a 'risk'. Trends Immunol 2002 Nov;23(11):530-534.
Gee JM, Kalil A, Shea C, Becker KJ. Lymphocytes – Potential mediators of postischemic injury and neuroprotection. Stroke 2007 Feb;38(2):783-788.
Louveau A, Smirnov I, Keyes TJ, et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015 Jul 16;523(7560):337-341.
Aspelund A, Antila S, Proulx ST, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med 2015 Jun 29;212(7):991-999.
Kitamura D, Roes J, Kuhn R, Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature 1991 Apr 4;350(6317):423-426.
Ren XF, Akiyoshi K, Dziennis S, et al. Regulatory B cells limit CNS inflammation and neurologic deficits in murine experimental stroke. J Neurosci 2011 Jun 8;31(23):8556-8563.
Bodhankar S, Chen Y, Vandenbark AA, Murphy SJ, Offner H. Treatment of experimental stroke with IL-10-producing B-cells reduces infarct size and peripheral and CNS inflammation in wild-type B-cell-sufficient mice. Metab Brain Dis 2014 Mar;29(1):59-73.
Koutrolos M, Berer K, Kawakami N, Wekerle H, Krishnamoorthy G. Treg cells mediate recovery from EAE by controlling effector T cell proliferation and motility in the CNS. Acta Neuropathol Commun 2014;2:163.
Chavele KM, Ehrenstein MR. Regulatory T-cells in systemic lupus erythematosus and rheumatoid arthritis. FEBS Lett 2011 Dec 1;585(23):3603-3610.
Bodhankar S, Chen Y, Vandenbark AA, Murphy SJ, Offner H. IL-10-producing B-cells limit CNS inflammation and infarct volume in experimental stroke. Metab Brain Dis 2013 Sep;28(3):375-386.
Putheti P, Soderstrom M, Link H, Huang YM. Effect of glatiramer acetate (Copaxone) on CD4+CD25high T regulatory cells and their IL-10 production in multiple sclerosis. J Neuroimmunol 2003 Nov;144(1-2):125-131.
Ibarra A, Avendano H, Cruz Y. Copolymer-1 (Cop-1) improves neurological recovery after middle cerebral artery occlusion in rats. Neurosci Lett 2007 Sep 25;425(2):110-113.
Poittevin M, Deroide N, Azibani F, et al. Glatiramer Acetate administration does not reduce damage after cerebral ischemia in mice. J Neuroimmunol 2013 Jan 15;254(1-2):55-62.
Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation 2006 May 2;113(17):2105-2112.
Kleinschnitz C, Schwab N, Kraft P, et al. Early detrimental T-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood 2010 May 6;115(18):3835-3842.
Barba R, Martinez-Espinosa S, Rodriguez-Garcia E, Pondal M, Vivancos J, Del Ser T. Poststroke dementia: clinical features and risk factors. Stroke 2000 Jul;31(7):1494-1501.
Leys D, Henon H, Mackowiak-Cordoliani MA, Pasquier F. Poststroke dementia. Lancet Neurol 2005 Nov;4(11):752-759.
Bejot Y, Aboa-Eboule C, Durier J, et al. Prevalence of early dementia after first-ever stroke: a 24-year population-based study. Stroke 2011 Mar;42(3):607-612.
Doyle KP, Quach LN, Sole M, et al. B-lymphocyte-mediated delayed cognitive impairment following stroke. J Neurosci 2015 Feb 4;35(5):2133-2145.
Savva GM, Stephan BC, Alzheimer's Society Vascular Dementia Systematic Review G. Epidemiological studies of the effect of stroke on incident dementia: a systematic review. Stroke 2010 Jan;41(1):e41-e46.
Pruss H, Iggena D, Baldinger T, et al. Evidence of intrathecal immunoglobulin synthesis in stroke: a cohort study. Arch Neurol 2012 Jun;69(6):714-717.
Rostrom B, Link H. Oligoclonal immunoglobulins in cerebrospinal-fluid in acute cerebrovascular-disease. Neurology 1981;31(5):590-596.
Tsementzis SA, Chao SW, Hitchcock ER, Gill JS, Beevers DG. Oligoclonal immunoglobulin-G in acute subarachnoid hemorrhage and stroke. Neurology 1986 Mar;36(3):395-397.
Zhang Y, Da RR, Guo W, et al. Axon reactive B cells clonally expanded in the cerebrospinal fluid of patients with multiple sclerosis. J Clin Immunol 2005 May;25(3):254-264.
Offner H, Hurn PD. A novel hypothesis: regulatory b lymphocytes shape outcome from experimental stroke. Transl Stroke Res 2012 Sep 1;3(3):324-330.
Fu Y, Liu Q, Anrather J, Shi FD. Immune interventions in stroke. Nat Rev Neurol 2015 Sep;11:524-535.
Warnke C, Stettner M, Lehmensiek V, et al. Natalizumab exerts a suppressive effect on surrogates of B cell function in blood and CSF. Mult Scler 2015 Jul;21(8):1036-1044.
Fu Y, Zhang N, Ren L, et al. Impact of an immune modulator fingolimod on acute ischemic stroke. Proc Natl Acad Sci U S A 2014 Dec 23;111(51):18315-18320.
Fu Y, Hao J, Zhang N, et al. Fingolimod for the treatment of intracerebral hemorrhage: a 2-arm proof-of-concept study. JAMA Neurol 2014 Sep;71(9):1092-1101.
Nakamura M, Matsuoka T, Chihara N, et al. Differential effects of fingolimod on B-cell populations in multiple sclerosis. Mult Scler 2014 Sep;20(10):1371-1380.
Grutzke B, Hucke S, Gross CC, et al. Fingolimod treatment promotes regulatory phenotype and function of B cells. Ann Clin Transl Neurol 2015 Feb;2(2):119-130.
Inacio AR, Liu Y, Clausen BH, et al. Endogenous IFN-beta signaling exerts anti-inflammatory actions in experimentally induced focal cerebral ischemia. J Neuroinflammation 2015;12:211.
Carrasco YR, Fleire SJ, Cameron T, Dustin ML, Batista FD. LFA-1/ICAM-1 interaction lowers the threshold of B cell activation by facilitating B cell adhesion and synapse formation. Immunity 2004 May;20(5):589-599.
Yuseff MI, Pierobon P, Reversat A, Lennon-Dumenil AM. How B cells capture, process and present antigens: a crucial role for cell polarity. Nat Rev Immunol 2013 Jul;13(7):475-486.
Doll DN, Barr TL, Simpkins JW. Cytokines: their role in stroke and potential use as biomarkers and therapeutic targets. Aging Dis 2014 Oct;5(5):294-306.
Li G, Liang X, Lotze MT. HMGB1: the central cytokine for all lymphoid cells. Front Immunol 2013;4:68.
Avalos AM, Kiefer K, Tian J, et al. RAGE-independent autoreactive B cell activation in response to chromatin and HMGB1/DNA immune complexes. Autoimmunity 2010 Feb;43(1):103-110.
Kazmierski R, Guzik P, Ambrosius W, Kozubski W. [Leukocytosis in the first day of acute ischemic stroke as a prognostic factor of disease progression]. Wiad Lek 2001;54(3-4):143-151.
Furlan JC, Vergouwen MD, Fang J, Silver FL. White blood cell count is an independent predictor of outcomes after acute ischaemic stroke. Eur J Neurol 2014 Feb;21(2):215-222.
Hug A, Dalpke A, Wieczorek N, et al. Infarct volume is a major determiner of post-stroke immune cell function and susceptibility to infection. Stroke 2009 Oct;40(10):3226-3232.
Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry 2009 May 1;65(9):732-741.
Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity 2008 May;28(5):639-650.
Castillo-Trivino T, Braithwaite D, Bacchetti P, Waubant E. Rituximab in relapsing and progressive forms of multiple sclerosis: a systematic review. PLoS One 2013;8(7):e66308.
Hawker K, O'Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol 2009 Oct;66(4):460-471.
Tuzun E, Zhou L, Baehring JM, Bannykh S, Rosenfeld MR, Dalmau J. Evidence for antibody-mediated pathogenesis in anti-NMDAR encephalitis associated with ovarian teratoma. Acta Neuropathol 2009 Dec;118(6):737-743.
Dalmau J. Status epilepticus due to paraneoplastic and nonparaneoplastic encephalitides. Epilepsia 2009 Dec;50(Suppl. 12):58-60.
Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. The Lancet Neurol 2011 Jan;10(1):63-74.
Kashyape P, Taylor E, Ng J, Krishnakumar D, Kirkham F, Whitney A. Successful treatment of two paediatric cases of anti-NMDA receptor encephalitis with cyclophosphamide: the need for early aggressive immunotherapy in tumour negative paediatric patients. Eur J Paediatr Neurol 2012 Jan;16(1):74-78.
Acknowledgments
This work was supported by American Heart Association 14SDG18410020 and NIH NS088555.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
U.M.S. and K.P. should be considered co-first authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1224 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Selvaraj, U.M., Poinsatte, K., Torres, V. et al. Heterogeneity of B Cell Functions in Stroke-Related Risk, Prevention, Injury, and Repair. Neurotherapeutics 13, 729–747 (2016). https://doi.org/10.1007/s13311-016-0460-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-016-0460-4