
Abstract
Neuromyelitis optica spectrum disorders (NMOSD) are important evolving entities, which have reached much attention in the recent years. NMOSD are characterized by inflammatory lesions in the optic nerves, spinal cord, and central parts of the brain, as well as an autoimmune process directed against aquaporin-4. As disability in NMOSD accumulates by inflammatory damage from attacks, both the treatment and prevention of attacks are decisive for the long-term outcome. NMOSD attacks are treated with high-dose intravenous corticosteroids and apheresis therapies, in particular therapeutic plasma exchange. In cases of incomplete remission, escalation of attack treatment is recommended. Preventive therapy is immunosuppressive and should by commenced as early as possible. Apart from classical immunosuppressants such as azathioprine and mycophenolate mofetil, repurposed biologicals are increasingly used. B-cell depletion with rituximab and other agents, inhibition of the interleukin-6 receptor with tocilizumab, and blockade of complement-mediated damage by eculizumab all are promising therapeutic strategies evaluated in randomized controlled trials. In this review, we will discuss present and future immunotherapies for NMOSD and also consider combination of treatments, plasma, cellular and other therapies. Current advances in immunopathological knowledge are translated into innovative concepts and begin a new era of NMOSD therapy.
Similar content being viewed by others
Introduction
Neuromyelitis optica (NMO) and NMO spectrum disorders (NMOSD) have been recognized in recent years as a distinguished disease entity owing mainly to identification of pathogenic autoantibodies directed against the astrocytic water channel aquaporin-4 (AQP4-IgG) [1–4]. Although being rare diseases with an incidence and prevalence of 0.05–0.40 and 0.52–4.40 per 100,000, respectively, NMO and NMOSD occur globally in various ethnicities [5], and often pose a high disease burden for affected individuals.
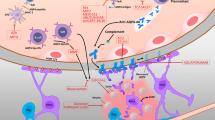
The autoimmune neuroinflammation of NMO and NMOSD involves the spinal cord, typically as longitudinally extensive transverse myelitis (LETM), the optic nerves and optic chiasm, and central parts of the brain. Apart from optic nerve/optic chiasm and spinal cord lesions, magnetic resonance imaging (MRI) shows cerebral lesions in a large proportion of patients, which might present as small and localized or extensive hemispheric lesions, periependymal lesions surrounding the aqueduct, the third and fourth ventricles, and brain stem lesions [6]. Barkhof criteria may sometimes be fulfilled, but generally lesions have an atypical appearance or location for multiple sclerosis (MS), the main differential diagnosis. Several clinical features highly distinctive for NMO and NMOSD have been identified, which include simultaneous LETM and optic neuritis (ON), bilateral ON, and intractable nausea with hiccups and vomiting [7]. Besides clinical evaluation, AQP4-IgG serology and MRI, cerebrospinal fluid diagnostics, evoked potentials, and optical coherence tomography guide diagnosis of NMO and NMOSD [8].
While ON and LETM are the classical hallmarks of NMO, the spectrum of NMO constantly expanded in recent years [9]. It includes AQP4-IgG-positive patients with isolated ON or LETM, both monophasic or relapsing, with isolated cerebral lesions involving the abovementioned locations, with 1 or several short myelitis lesions [10], or with combinations thereof. Furthermore, NMO and NMOSD can be associated with myopathy and occur in the context of systemic autoimmune disease or cancer [9]. The case for AQP4-IgG-negative NMO is more complex, as these patients form a heterogeneous group with demographic and clinical characteristics different from seropositive patients [11]. Autoantibodies targeting the myelin oligodendrocyte glycoprotein have been reported in some AQP4-IgG-negative patients with a classical clinical presentation of NMO [12, 13], however, anti-myelin oligodendrocyte glycoprotein-antibodies are not specific for NMO and occur in a variety of other demyelinating diseases [14]. Recently, new diagnostic criteria were published that present a uniform concept combining NMO and NMOSD [15], which is why the term “NMOSD” will be used throughout this review. As for MS after definition of the McDonald criteria it is expected that the new diagnostic criteria for NMOSD will lead to an increase in patient numbers, particularly in AQP4-IgG-negative patients.
It was demonstrated long ago that immunosuppressive therapy with corticosteroids is beneficial to induce remission from acute exacerbations and for long-term treatment of NMOSD [16]. Azathioprine (AZA) was among the first immunosuppressants reported to be effective for NMOSD [17]. Subsequently, the new disease-modifying drugs approved for MS were given for NMOSD, with variable success [18–20], while more promising results came from B-cell depletion therapy [21]. The latter approach was a consequence of pathoanatomical and serological studies identifying humoral disease mechanisms, particularly complement activation and NMO-IgG, involved in the pathogenesis of NMOSD [22, 23].
Several national and international guidelines describing diagnosis and treatment of NMOSD have been published [8, 24–27], yet most therapeutic recommendations are still expert-based because until recently all studies reporting treatment outcomes had been conducted in a nonrandomized and often retrospective setting.
With advances in knowledge about the immunopathology of NMOSD [28], innovative therapeutic concepts [29], and the first randomized controlled trials coming up, we are now approaching a new era of NMOSD therapy. In this review, we will discuss present and future immunotherapies for NMOSD. For symptomatic treatments, which are similar to those applied in MS, we refer to respective reviews [30, 31].
Treatment of NMOSD Attacks
Unlike MS, NMOSD almost never take a progressive disease course and disability is accumulated by inflammatory damage from attacks. Only every 2. to 5. NMOSD attack shows complete recovery [11, 32]. In particular, bilateral ON and myelitis have low remission rates [33]. As successful treatment of attacks determines long-term outcome and disability, NMOSD attacks should be treated “hard and early” and escalation of therapy is recommended. In a retrospective study of 871 NMOSD attacks, escalation of attack therapy significantly improved outcome; after the first treatment course 19.1 % of attacks showed complete remission and 16.4 % no remission, and after the last treatment course 21.6 % showed complete remission and 6.0 % no remission [33].
Recommendations for the treatment of NMOSD attacks were adapted from studies of MS and idiopathic ON. No controlled trials have been performed in NMOSD and most studies examining apheresis therapies have reported outcomes of both entities.
Acute exacerbations of NMOSD are usually treated with high-dose intravenous (IV) methylprednisolone (IVMP) at a dose of 1000 mg for 3–5 days with or without oral tapering [8, 24–26]. It has been shown that each, a first, second, and third course of IVMP significantly improves clinical disability in patients with MS and NMOSD [34], and that early administration is associated with preservation of the retinal nerve fiber layer thickness in NMOSD [35].
When remission is absent or insufficient, therapeutic plasma exchange (TPE; 5–7 cycles) should be used [8, 24–26]. The efficacy of TPE for relapsing–remitting MS and other inflammatory demyelinating diseases was demonstrated in a randomized, controlled trial and several uncontrolled studies [36–40]. TPE was also reported to be beneficial for NMOSD attacks [40–43]. TPE effectively eliminates pathogenic AQP4-IgG from the patient’s circulation [44], which is one reason to use TPE in NMOSD. However, TPE improves attack-related disability not only in seropositive but also in seronegative patients [40, 42, 45], where putatively other types of autoantibodies may exist. Attacks other than ON, fewer prior attacks, shorter disease duration, and lower pre-existing disability are factors associated with the favorable outcome of TPE [40, 43–45]. If TPE is not applicable, for example, owing to contraindications, a second course of IVMP at a higher dose of up to 2 g for 5 days can be given [8].
In some countries, immunoadsorption (IA), a method to remove immunoglobulins from the circulation by adsorption to either tryptophan or protein A, is used as an alternative to TPE for the treatment of exacerbations of MS and NMOSD [46, 47]. Several adult and pediatric cases have been reported in whom NMOSD attacks were successfully treated with IA [33, 46, 48–50].
Although mostly given as escalation therapies, TPE and IA might also be used as initial treatment for severe attacks, in particular when previous attacks have responded well to apheresis therapies but not to IVMP [8]. Interestingly, 3 reports have suggested that TPE in parallel with IVMP is more effective in reaching remission from NMOSD attacks than IVMP alone [32, 42, 43]. In line with these results, a retrospective analysis of 871 NMOSD attacks revealed that first-line therapy with PE or IA is superior to IVMP when the spinal cord is affected [33].
Resistant cases without improvement after ≥ 1 course of both IVMP and apheresis therapies have occasionally been escalated to early highly active immunotherapy such as IV cyclophosphamide [26], mitoxantrone [8], or tocilizumab [51, 52], which induced a dramatic improvement in disability in single patients.
Inhibition of complement-mediated damage, for example by IV immunoglobulins (IVIG), eculizumab, or C1-esterase inhibition, might be another mechanism to induce remission from refractory NMOSD attacks. A retrospective study examining IVIG as an attack therapy in patients nonresponsive to IVMP with or without TPE revealed beneficial effects in 5/11 attacks [53]. Two randomized controlled trials are currently evaluating the efficacy of IVIG for NMOSD attacks. Recently, the C1-esterase inhibitor Cinryze (ViroPharma, Exton, PA, USA) was given as add-on therapy to IVMP in an open-label study of 10 patients with AQP4-IgG-positive NMOSD [54]. Cinryze therapy was safe and associated with disability improvement to preattack levels in 9/10 patients after 1 month. Finally, inhibition of neutrophil elastase with sivelestat was beneficial in animal models of NMOSD [55, 56], and a small open-label trial evaluating sivelestat for NMOSD attacks is currently being conducted in Japan.
Prevention of NMOSD Attacks
General Considerations
As attacks are decisive for the long-term outcome of NMOSD, both prevention of attacks and attenuation of attack severity are important treatment goals. Most patients with NMOSD have a relapsing disease course, and the median time between first and second attack is < 1 year [11]. Natural history cohorts describing the annual relapse rate (ARR) in untreated patients do not exist for NMOSD; however, it is noteworthy that AQP4-IgG-positive patients with late-onset have attacks even in the oldest age categories [57].
There are probably no differences in relapse rates and clinical features between AQP4-IgG-positive patients with classical NMO and NMOSD, but AQP4-IgG-negative patients more often have bilateral ON or simultaneous ON and myelitis at disease onset than AQP4-IgG-positive patients [11, 58]. While 1 study reported that a monophasic disease course is more common in AQP4-IgG-negative patients [11], this was not found in another study [58]. It is unclear whether AQP4-IgG seronegative and seropositive NMOSD are driven by similar humoral immune mechanisms [29]. Therapeutic recommendations for AQP4-IgG-positive NMOSD cannot bona fide be transferred to seronegative patients, who should be treated on an individual basis, guided by severity and remission of the first attack and the clinical course [8]. In patients diagnosed with AQP4-IgG-positive NMOSD, however, preventive therapy should be started as soon as diagnosis is confirmed [8].
Several immunomodulatory drugs approved for MS were shown to exacerbate the disease course in AQP4-IgG-positive NMOSD. Although only assessed by retrospective studies, the marked worsening of disability reported in some patients treated with interferon-β [19, 20, 59–62], natalizumab [63–65], and fingolimod [66, 67] should prompt us to avoid these therapies. Alemtuzumab, a T- and B-cell-depleting agent, was also shown to exacerbate NMOSD in single patients [51, 68, 69]. At present, there are insufficient data to support or discourage use of glatiramer acetate, teriflunomide, and dimethylfumarate in NMOSD [8].
Although not investigated by randomized, controlled trials, it is now well accepted that treatment for the prevention of NMOSD attacks should be immunosuppressive [8, 24–26]. Several classical immunosuppressants and some repurposed biologicals have been recommended for NMOSD and will be discussed below. Ambiguous cases of autoimmune neuroinflammation not fulfilling criteria for MS or NMOSD are often treated with broad-acting immunosuppressive drugs such as AZA or methotrexate (MTX) [70].
Before starting an immunosuppressive or biological therapy, patients must be informed about the respective risks and potential complications, such as infections, cytotoxicity and myelotoxicity, malignancy, infertility, and, in women, the need for contraception. Pregnancy and chronic infections, particularly HIV, hepatitis B and C, and tuberculosis, should be excluded prior to start of therapy.
The duration of prophylactic immunotherapy in NMOSD is not well defined [26]. Periods of increased attack frequency can be followed by relapse-free times for years before disease activity unpredictably recurs. It has been suggested to continue immunosuppressive therapy for at least 5 years after a first LETM attack [71]. However, any attempt to reduce or stop immunotherapy for NMOSD has to be discussed carefully with the patient, balancing the benefits and risks of the applied therapies [26].
Classical Immunosuppressants
Oral Corticosteroids
Corticosteroids bind to the glucocorticosteroid receptor expressed on all cells and induce a wide variety of genomic changes that lead to immunosuppression [72]. Oral low-dose corticosteroids (usually prednisolone, up to 1 mg/kg/day; Table 1) are used for NMOSD to taper out steroids after IVMP attack therapy, in combination with other immunosuppressants, particularly during dosing in and with breakthrough disease, and sometimes as monotherapy. A small retrospective study of 9 patients with NMOSD reported that the median ARR decreased from 1.48 in untreated patients to 0.49 in corticosteroid-treated patients; doses >10 mg/day were associated with fewer attacks [73]. Long-term use of oral corticosteroids above ~7.5 mg/day is associated with many side effects, for example hyperglycemia, hypertension, weight gain, and osteoporosis, and therefore should be avoided as monotherapy for NMOSD.
AZA
The purine synthesis inhibitor AZA preferentially affects proliferation of B and T cells and is the most widely studied immunosuppressant for NMOSD. Several studies including a total of almost 400 patients have shown that AZA reduces the ARR, sometimes associated with improvement of neurological disability [17, 74–80]. A retrospective study evaluating classical NMO and patients with AQP4-IgG-positive NMOSD reported a decrease in the mean ARR from 2.18 to 0.64 in 70 patients treated with AZA for > 1 year, with or without concomitant corticosteroid therapy [77]. A reduced dose of < 2 mg/kg/day AZA was associated with more, and an increase of the mean corpuscular volume by at least 5 fl from baseline with fewer relapses (both to be confirmed in further studies). Clinical disability measured by the expanded disability status scale (EDSS) and visual acuity improved or remained unchanged in 61 % and worsened in 39 % of patients with > 12 months of follow-up on AZA therapy. AZA was discontinued in 38/99 patients, mostly for side effects and lack of efficacy. Another retrospective study of 103 patients with AQP4-IgG-positive NMOSD confirmed these findings, revealed a reduction of the median ARR in 89 % of patients (median of 1.5 pretreatment to 0 on AZA), and an improvement or stabilization of the EDSS in 78 % of patients, but again had a high discontinuation rate of AZA (46 %) [78]. A third retrospective study of 31 patients with AQP4-IgG-positive or AQP4-IgG-negative NMOSD described a reduction in the mean ARR from 2.26 to 0.63 with a failure rate in 53 % of patients, despite concomitant prednisone therapy [79]. Another retrospective study of AZA plus long-term low-dose corticosteroid therapy in 77 Chinese patients found a relapse-free status at median follow-up of 19 months in 44 patients (57.1 %) and significant improvements of the ARR, EDSS, and modified Rankin Scale [80].
While the discussed studies have shown that AZA can reduce the ARR in patients with NMOSD, 2 recent retrospective studies suggested that the risk for further NMOSD attacks was significantly higher with AZA treatment than with B-cell-depleting rituximab therapy [hazard ratio (HR) 2.12 and 1.82, respectively] [79, 81], and that severe attacks occurred more often in patients treated with AZA (HR 11.66) [81].
AZA is given at a dose of 2–3 mg/kg/day, should be slowly tapered in with monitoring of hematological parameters and transaminases, and reaches clinical activity after 3–6 months. Prior to starting AZA, testing for thiopurine methyltransferase enzyme activity may be useful as patients with low activity are of higher risk for severe side effects [8, 26]. We suggest adjusting the target dose of AZA by the total lymphocyte count, which should decrease to 600–1000/μl, the leukocyte count, which should remain at > 3000/μl, and the mean corpuscular volume, which should increase by about 5 % from baseline. A correlation between the dosage of AZA and clinical efficacy was reported for NMOSD and other indications [77, 82]. Regular differential blood cell count and transaminase monitoring are mandatory. Side effects of AZA include nausea, diarrhea, fatigue, leuko- and lymphopenia, infections, liver toxicity, bone marrow suppression, and malignancies with long-term use.
Mycophenolate Mofetil
Mycophenolate mofetil (MMF) is a prodrug of the active metabolite mycophenolic acid. It inhibits lymphocyte proliferation by suppression of guanosine nucleotide biosynthesis and is used for psoriasis, proliferative lupus nephritis, and renal transplant rejection. Four retrospective cohorts evaluating 24–58 patients with mostly AQP4-IgG-positive NMOSD have reported beneficial outcomes of MMF therapy with or without concomitant corticosteroids [79, 81, 83, 84]. MMF treatment for a median of 20–27 months resulted in a reduction of the ARR by 80–93 % and improvement of the median EDSS in 3/4 studies (not reported in 1). More than 90 % of patients had stable or improved disability [83, 84], and 46–65 % were relapse-free [79, 81, 83, 84]. Discontinuation rates due to disease activity or side effects were 15–25 %.
In comparison with B-cell-depleting rituximab therapy there was no significant difference for the risk of further NMOSD attacks (HR 1.48 and 1.27, respectively) [79, 81], although 1 study found that severe attacks occurred significantly more often with MMF than with rituximab (HR 5.96) [81].
MMF is tapered in with a target dose of 1500–3000 mg/day, guided by the total lymphocyte count, which should decrease to 1000–1500/μl, but also following a plasma trough level of 1–2 µg/ml in neuroimmunological indications. The treatment effect usually occurs within 2–3 months. Monitoring of the differential blood cell count and liver function is mandatory. Gastrointestinal complications, hypercholesterolemia, elevation of transaminases, myelotoxicity, and infections, including progressive multifocal leukoencephalopathy, are potential side effects of MMF therapy.
MTX
MTX, a folic acid antagonist, is used for hematological malignancies, rheumatoid arthritis, psoriasis, myasthenia gravis, and other autoimmune diseases. Kitley et al. [85] treated 14 patients with AQP4-IgG-positive NMOSD for a median of 21.5 months with 17.5 mg MTX (median). The ARR was reduced by 87 %; 79 % of patients had a stable or improved EDSS; 43 % were relapse-free. However, 13/14 patients had received concomitant immunotherapy, 11 with oral prednisolone, 1 with rituximab, and 1 with tacrolimus. In another retrospective study of 9 patients with NMOSD, 17.5 mg MTX accompanied by low-dose prednisone therapy reduced the ARR by 64 %, 2 patients were relapse-free, and 5 had a stable or improved EDSS [86]. In 3 patients, MTX was used as a de-escalation strategy after 6 months of cyclophosphamide therapy; however, 2 of these 3 patients had further attacks while on MTX. An older, less well-documented study revealed stable disease (without new clinical or MRI events) and improvement of EDSS over a period of 24 months in all 7 patients with Devic disease who were treated with a combination of 50 mg IV MTX weekly and prednisone 1 mg/kg/day (with subsequent tapering) [87]. MTX was generally well tolerated in patients with NMOSD [85–87].
MTX is tapered in and folic acid supplementation is recommended. As for other immunosuppressants regular differential blood cell count and transaminases should be monitored and infections anticipated.
Ciclosporin A and Tacrolimus
The calcineurin inhibitors ciclosporin A and tacrolimus are frequently used for patients with various autoimmune diseases or to prevent organ transplant rejection. Kageyama et al. [88] described 9 patients with AQP4-IgG-positive NMOSD treated with a combination of ciclosporine A (median dose 150 mg/day) and prednisone for 13–51 months, which improved the ARR in 8 patients (3 were relapse-free, mean ARR reduction 86 %) and stabilized the EDSS in 7.
Tacrolimus (1–6 mg/day) was given as add-on therapy to prednisolone (median initially 13 mg/day, after tapering 1.5 mg/day) in 25 Japanese patients with NMOSD [89]. Twenty-three patients (92 %) remained relapse-free in a treatment time of 2–44 months (median 12 months) and 2 patients had side effects (diabetes and cholangitis). Another patient with AQP4-IgG-positive NMOSD associated with Sjögren syndrome and failure of intravenous cyclophosphamide was stable for 3 years with tacrolimus [90], while in 1 patient with systemic lupus erythematosus-associated NMOSD tacrolimus was not effective [91].
Mitoxantrone
Mitoxantrone is an antineoplastic topoisomerase II inhibitor that inhibits both DNA and RNA synthesis and suppresses T- and B-cell immunity. Two recent observational studies of 20 and 51 patients with NMOSD found a reduction in ARR of 75 % and 80 % and a relapse-free status in the first year of treatment of 50 % and 70 %, respectively, during mitoxantrone treatment (duration up to 22 months, cumulative dose up to 120 mg/m2) [92, 93]. In one study all patients were AQP-IgG-positive [92]; in the other AQP4-IgG-positive patients had more relapses than AQP4-IgG-negative patients [93]. Another small open-label study of 5 patients with NMOSD demonstrated beneficial effects of mitoxantrone on disability and MRI in 4 patients, with 3 patients staying relapse-free [94].
Mitoxantrone is usually started as induction therapy with 3 monthly pulses of 12 mg/m2 body surface area supplemented with 1 g IVMP, followed by further infusions every 3 months until the maximum dose of 100 mg/m2 (120 mg/m2 in exceptional cases) is reached. Different dosing regimens exist. Cardiac function and differential blood cell count have to be monitored prior each infusion. Decrease in left ventricular ejection fraction and hematological malignancies occur as dose-dependent effects in some patients and limit the use of mitoxantrone to severe cases of NMOSD.
Cyclophosphamide
Immunoablative or immunosuppressive therapy with IV cyclophosphamide was reported to induce stable disease in single patients with NMOSD, often in the context of systemic lupus erythematosus [91, 95]. However, 3 retrospective studies of 4, 5, and 7 patients, respectively, revealed breakthrough disease of NMOSD with IV cyclophosphamide therapy in all but one patient each [87, 96, 97]. Therefore, IV cyclophosphamide is only recommended when other therapies fail or are not available [8].
Combination Therapies of Immunosuppressants
Immunosuppressants are often combined with oral corticosteroids, in particular during tapering in and in patients not responding to monotherapy. Adjunctive therapy of AZA [17, 75, 77, 79], MMF [79, 83], MTX [85–87], ciclosporine A [88], and tacrolimus [89] with oral corticosteroids have all been reported to be (partly) effective and safe in NMOSD. In breakthrough disease intermittent TPE might be added to immunosuppressive treatment [8, 98], but usually escalation to a biological is preferable.
Biologicals
After recovering the importance of humoral immune mechanisms for NMOSD [22, 23], soon more specific therapies aiming to suppress either B cells, plasma cells, or downstream effector mechanisms were advocated. Nowadays, a couple of approaches derived from basic research data are being translated into clinical practice and biologicals are about to become the mainstay of NMOSD therapy.
Rituximab and Other B-cell-depleting Therapies
Rituximab is a chimeric anti-CD20 monoclonal antibody that depletes both naïve and memory B cells. It was the first specific immunosuppressant used for NMOSD. The pivotal open-label study from Cree et al. [21] established B-cell depletion as a therapeutic principle for NMOSD; 6/8 patients were relapse-free after 1 year of treatment. Several clinical case series and retrospective analyses (including 10–55 patients each, treatment up to 8 years) have confirmed these findings and reported a reduction of ARR in 87–96 %, freedom from relapses in 44–72 %, and an improvement of disability in 80–100 % of patients [79, 81, 97, 99–102]. Most patients in these studies were AQP4-IgG-positive, but response rates in patients with AQP4-IgG-negative NMOSD seem to be similar [97, 102]. Treatment responses to rituximab appear to be more favorable than for classical immunosuppressive therapies [79, 81, 97], but caution has to be applied as all reported studies are prone to different kinds of bias owing to their retrospective design. Although previously often used as escalation therapy, its high efficacy makes rituximab an option for first-line treatment of highly active NMOSD [8].
There are various treatment regimens for rituximab in NMOSD, yet it has not been identified which has the optimal ratio of efficacy, side effects, and costs. Usually, rituximab infusions are applied either at a dosage of 2 treatments of 1 g with a 2-week interval or, for oncological indications, 4 times per 375 mg/m2 body surface area with weekly intervals. More than 80 % of patients remain B-cell depleted 6 months after rituximab treatment [103], and re-dosing is often done at regular 6-month intervals [79], which however runs the risk that B cells in some patients are already repopulating. Another approach is to monitor B-cell counts closely and to re-dose rituximab before recurrence of circulating B cells (CD19+ or CD20+ B cells > 0.1 % of total lymphocytes, CD27+ memory B cells > 0.05 % of peripheral blood mononuclear cells) [99]. A genetic polymorphism in the fragment c gamma receptor 3A (FCGR3A) allele has been shown to be associated with insufficient memory B-cell depletion and an elevated risk of attacks during rituximab treatment [104]. It is controversial whether a reduction in the dose during long-term treatment or even lower initial doses of rituximab are sufficient to suppress disease activity [100, 103, 105, 106]. Generally, optimal dosing of rituximab is important to lower the failure rate [79, 103]. Relapses occurring in the setting of (incipient) B-cell repopulation should not be regarded as a nonresponse to rituximab but rather as dosing failure.
Moreover, relapses were reported to occur in the first weeks after the first rituximab application, probably owing to transient elevation of proinflammatory cytokines such as tumor necrosis factor-α, interleukin (IL)-6, and B-cell activating factor, all of which promote maturation and survival of B cells and plasmablasts [107, 108]. In line with these findings AQP4-IgG transiently increase after initiation of rituximab, with a subsequent decrease [107]. Lower titers of AQP4-IgG are probably associated with less disease activity but have not yet been established as a reliable biomarker of treatment response in rituximab-treated patients [74, 100, 101]. It is unknown whether the clinical effects of B-cell depletion are mediated by a reduction of AQP4-IgG or by inhibition of other proinflammatory B-cell functions. Notably, rituximab does not deplete antibody-producing plasma cells.
Despite the apparent success of rituximab in NMOSD it has to be stated that none of the abovementioned studies was controlled with a placebo arm or active comparator, making assessment of the real therapeutic effect difficult. In this regard it is of great importance that a randomized controlled trial was started, which evaluates the efficacy of a novel anti-CD19 humanized monoclonal antibody (MEDI-551) against placebo to prevent NMOSD attacks (Table 2) [109]. Further B-cell-depleting therapies targeting CD19 or CD20 are being developed for MS and other autoimmune diseases, for example ocrelizumab and ofatumumab, all of which potentially could be used for NMOSD in the future. A phase I trial with ublituximab (anti-CD20 antibody) to treat acute NMOSD attacks is in preparation [110].
Although rituximab generally is well tolerated, patients receiving this drug or other B-cell-depleting monoclonal antibodies should be monitored closely for allergic reactions (preventive premedication is obligatory), infections, low IgG/IgM levels, and late-onset neutropenia [111, 112]. Progressive multifocal leukoencephalopathy is another potential complication, which has not yet been reported in patients with NMOSD or MS treated with rituximab. Vaccinations with inactivated vaccines are safe during rituximab therapy in patients with NMOSD, but a reduction in the immune response should be anticipated [113].
IL-6 Receptor Inhibition
Another method to target the humorally mediated immune process of NMOSD is inhibition of the IL-6/IL-6 receptor axis [114]. IL-6 is produced by stromal, epithelial, muscle, and immune cells, binds to either soluble or membrane-bound IL-6 receptor, and mediates its pleotropic effects through the transmembrane protein gp130 [115]. It has multiple functions in the immune system, including promotion of T- and B-cell activation, T helper 17 differentiation, and plasmablast survival.
Increased levels of IL-6 were detected in the serum and cerebrospinal fluid of patients with NMOSD, which correlated with disease activity and AQP4-IgG titers [116, 117]. Production and secretion of AQP4-IgG by B-cell-derived plasmablasts was shown to be dependent on IL-6, and blockade of IL-6 receptor signaling by an anti-IL-6 receptor antibody reduced the survival of plasmablasts in vitro [118]. Subsequently, several case reports revealed a reduction of disease activity in patients with severe, AQP-IgG-positive NMOSD using the anti-IL-6 receptor antibody tocilizumab, which is approved for the treatment of rheumatoid arthritis (IV and subcutaneous application) [51, 119, 120].
The efficacy of tocilizumab is supported by the congruent results of 2 recent open-label studies reporting a total of 15 patients from Germany and Japan with AQP-IgG-positive NMOSD [121, 122]. Araki et al. [121] treated 6 females and 1 male with high disease activity (mean ARR 2.9 ± 1.1, mean EDSS 5.1 ± 1.7) monthly with 8 mg/kg tocilizumab as add-on therapy to either prednisolone, AZA, ciclosporin, or tacrolimus. During 12 months of therapy, both ARR (0.4 ± 0.8) and EDSS (4.1 ± 1.6) significantly decreased and fatigue improved. Ringelstein et al. [122] reported the disease course of 8 female patients with severe NMOSD nonresponsive to rituximab (median ARR 4.0, median EDSS 7.3), which significantly improved during monotherapy with tocilizumab (median ARR 0.4, median EDSS 5.5) for up to 4 years. Relapses were mostly mild and occurred either in the first 2.5 months of therapy or after a reduction of application frequency (>40 days) or dosage (6 mg/kg instead of 8 mg/kg). Signs of disease activity on MRI were absent in 7/8 patients and AQP4-IgG titers significantly dropped.
Interestingly, both studies reported a reduction of chronic pain and about half of the patients became free of pain with tocilizumab therapy. Neuropathic pain is a frequent and often intractable symptom of NMOSD [123, 124]. Among other pleiotropic effects of IL-6, direct modulation of nociceptive neurons is likely, because deletion of its signal transducing receptor gp130 in peripheral neurons significantly reduced pain in mice [125].
Infections, hypercholesterolemia, elevation of transaminases, and leukopenia are the most common adverse effects of tocilizumab, and were observed in patients with NMOSD [121, 122]. Possible tuberculosis reactivation and opportunistic infections make careful observations essential. As C-reactive protein is directly downregulated by tocilizumab, it cannot be used as a sensitive diagnostic marker.
The above results from nonrandomized studies suggest that inhibition of the IL-6/IL-6 receptor axis might be beneficial from early stages of the disease to prolonged treatment of severely affected patients. Apart from the reduction of ARR, tocilizumab might additionally improve disability and alleviate fatigue and chronic pain. Currently, tocilizumab represents an off-label therapy for NMOSD and was suggested as a third-line treatment for severe cases [8]. A new subcutaneous IL-6 receptor-blocking monoclonal antibody (SA 237; Chugai, Tokyo, Japan), which has a fourfold greater duration of action than tocilizumab, is currently being evaluated for NMOSD in 2 randomized controlled trials [126, 127]. Other monoclonal antibodies targeting IL-6 or the IL-6 receptor are being developed for non-neurological conditions.
Complement Inhibition
As membrane-bound AQP4-IgG activates the complement cascade, ultimately forming the membrane attack complex, astrocytes expressing the target antigen AQP4 are destroyed during the immune attack [128, 129]. Therefore, inhibition of complement activation should reduce damage to the central nervous system in patients with AQP4-IgG-positive NMOSD.
Eculizumab, a humanized monoclonal antibody targeting the complement protein C5, was approved as an orphan drug for paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Eculizumab inhibits the cleavage of C5 and prevents formation of the cytolytic terminal membrane attack complex C5b–C9. As the soluble peptide C5a has multiple roles in inflammation, including recruitment of basophils, macrophages, neutrophils, and lymphocytes to inflammatory sites, differentiation of T helper 1 cells, and interaction with Toll-like receptors [130, 131], upstream proinflammatory pathways might also be inhibited by eculizumab, potentially lowering attack frequency. An open-label pilot study investigated the efficacy and safety of eculizumab (600 mg IV weekly for 4 weeks, 900 mg in fifth week, then 900 mg every 2 weeks for 48 weeks) in 14 female patients with AQP4-IgG-positive NMOSD with high disease activity [132]. Eculizumab treatment reduced the median number of attacks from 3 to 0 and the median EDSS from 4.3 to 3.5. Twelve of 14 patients were relapse-free and disability stabilized or improved in all. Beside meningococcal sepsis with sterile meningitis, rheumatoid arthritis, and transient ischemic attack, in single patients each, no other serious adverse events occurred. While complement activity was suppressed during eculizumab treatment, AQP4-IgG titers remained unchanged. This interesting study demonstrates that complement-dependent cytotoxicity contributes to tissue damage in NMOSD and is a valuable target for both attack and preventive therapy. Nevertheless, infectious complications, particularly meningococcal meningitis (meningococcal vaccine is obligatory prior to the start of therapy), are an area of concern with complement-inhibiting therapies. The efficacy of eculizumab for AQP-IgG-positive NMOSD is currently being investigated in the randomized, controlled PREVENT trial [133].
Several other complement inhibitors, for example monoclonal antibodies targeting C1, and compounds inhibiting C1 esterase activity, C3, or C3a and C5a receptors, were assessed in preclinical models or have been suggested for theoretical reasons [29].
Combination Therapy with Biologicals
As commonly practiced in rheumatology, combination therapy of a biological with another immunosuppressive or immunomodulatory approach might be used in refractory NMOSD with high disease activity. Examples include combination of rituximab with MTX or IVIG [8], or tocilizumab with prednisolone, AZA, ciclosporin, or tacrolimus [121]. Care has to be taken as combination therapies implicate an increased risk of complications, particularly infections.
Plasma, Cellular and Other Therapies
Further empiric therapies are used or have been proposed for prevention of NMOSD attacks, some of which are summarized below.
First, plasma therapies aiming to reduce humoral factors, particularly circulating AQP4-IgG, cytokines and proteins of the complement system, may be beneficial for maintenance therapy of NMOSD. Apheresis therapies such as TPE are classically used to treat severe NMOSD attacks; however, TPE was also evaluated for prevention of attacks [98, 134]. Khatri et al. [134] reported 7 patients with AQP4-IgG-positive NMOSD who received 21–154 TPE treatments for a mean of 7.1 years concomitantly with either oral or IV corticosteroids, cyclophosphamide, or both. Initially, TPE was applied as induction therapy, followed by gradual tapering to once every 3–12 weeks. Only one patient was relapse-free, but the ARR was reduced in all patients and 5/7 patients had an improvement of disability. Although not reported so far, regular IA therapy may have a similar effect as maintenance therapy. Another plasma therapy, monthly IVIG, inhibited clinical attacks in single patients with NMOSD for treatment periods of up to 5.5 years [135, 136]. Therefore, in patients with contraindications for classical or selective immunosuppressants, particularly children, regular IVIG infusions might be tried [8].
Second, similar to other severe autoimmune diseases, cellular therapy, either as rescue hematopoietic stem cell therapy after immunoablative chemotherapy or as stem cell transplantation with immunoregulatory or regenerative function, was proposed for NMOSD. Greco et al. [137] reported improvement of clinical disability, cessation of attacks, and disappearance of AQP4-IgG in 2 patients with severe NMOSD after allogeneic hematopoietic stem cell transplantation. In contrast, 14/17 patients with NMOSD described in 2 studies and treated with autologous hematopoietic stem cell transplantation had further disease activity [138, 139]. Several registered clinical trials are underway to evaluate autologous mesenchymal or hematopoietic stem cell transplantation for NMOSD.
Third, granulocyte-, plasma cell-, cytokine-, or chemokine-directed therapies designed to modulate pathophysiologically relevant pathways are further potential treatments for NMOSD. Examples include antihistamines, bortezomib, and inhibitors of IL-8 or IL-17; however, these have not yet been investigated in the clinical setting.
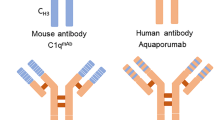
Finally, antigen-specific therapies with curative properties might be developed and are of particular interest, given that the autoantigen is known in NMOSD. One approach is to block competitively the binding of AQP4-IgG to AQP4 (e.g., by the nonpathogenic human monoclonal antibody aquaporumab); others include enzymatic deglycosylation or cleavage of AQP4-IgG [29]. Furthermore, various techniques to restore immune tolerance to AQP4 derived from animal models and other human autoimmune diseases have been suggested or are in (pre-)clinical development [29, 140]. Examples are DNA vaccination [141], T-cell-based vaccines [142], peptide-coupling strategies [143], and adoptive transfer of tolerogenic dendritic cells or regulatory T cells [144].
Conclusions
Treatment and prevention of attacks are equally important to control NMOSD, which often has an unfavorable prognosis. As the remission status of attacks determines long-term outcome and disability, aggressive therapy of NMOSD attacks is warranted. Apart from IVMP, TPE, and IA, which were derived from MS therapy and target cellular and humoral immune processes, inhibition of complement activation or other mechanisms relevant for disease pathogenesis might be helpful to treat NMOSD attacks. Approved immunosuppressive drugs and increasingly repurposed biologicals targeting specific immune processes are used for preventive therapy.
In contrast to MS, gradual progression of disability is uncommon in NMOSD, which should allow freedom of disease activity being reached simply through the prevention of attacks. Theoretically, the absence of attack-related inflammation could also release repair processes such as remyelination. Indeed, several studies investigating broad or selective immunosuppression reported an improvement of disability during the course of treatment [21, 81, 83, 84, 87, 92, 102, 121, 122].
A future goal is to transfer the current empiric treatment recommendations for NMOSD to an evidence based standard of care, which will only be possible by conducting randomized controlled trials [145]. First treatment trials are underway and will potentially broaden our therapeutic spectrum for NMOSD.
References
Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006;66:1485-1489.
Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol 2007;6:805-815.
Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 2005;202:473-477.
Jarius S, Wildemann B. AQP4 antibodies in neuromyelitis optica: diagnostic and pathogenetic relevance. Nat Rev Neurol 2010;6:383-392.
Pandit L, Asgari N, Apiwattanakul M, et al. Demographic and clinical features of neuromyelitis optica: A review. Mult Scler 2015;21:845-853.
Kim HJ, Paul F, Lana-Peixoto MA, et al. MRI characteristics of neuromyelitis optica spectrum disorder: an international update. Neurology. 2015;84:1165-1173.
Jurynczyk M, Craner M, Palace J. Overlapping CNS inflammatory diseases: differentiating features of NMO and MS. J Neurol Neurosurg Psychiatry 2015;86:20-25.
Trebst C, Jarius S, Berthele A, et al. Update on the diagnosis and treatment of neuromyelitis optica: recommendations of the Neuromyelitis Optica Study Group (NEMOS). J Neurol 2014;261:1-16.
Zekeridou A, Lennon VA. Aquaporin-4 autoimmunity. Neurol Neuroimmunol Neuroinflamm 2015;2:e110.
Flanagan EP, Weinshenker BG, Krecke KN, et al. Short myelitis lesions in aquaporin-4-IgG-positive neuromyelitis optica spectrum disorders. JAMA Neurol 2015;72:81-87.
Jarius S, Ruprecht K, Wildemann B, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: A multicentre study of 175 patients. J Neuroinflammation 2012;9:14.
Kitley J, Woodhall M, Waters P, et al. Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology 2012;79:1273-1277.
Sato DK, Callegaro D, Lana-Peixoto MA, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology 2014;82:474-481.
Mader S, Gredler V, Schanda K, et al. Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J Neuroinflammation 2011;8:184.
Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015;85:177-189.
Mueller W, Mortier R. Corticoidbehandlung der Neuromyelitis optica. Dtsch Z Nervenheilkd 1963;185:170-182 (in German).
Mandler RN, Ahmed W, Dencoff JE. Devic's neuromyelitis optica: a prospective study of seven patients treated with prednisone and azathioprine. Neurology 1998;51:1219-1220.
Bergamaschi R, Uggetti C, Tonietti S, Egitto MG, Cosi V. A case of relapsing neuromyelitis optica treated with glatiramer acetate. J Neurol 2003;250:359-361.
Papeix C, Vidal JS, de Seze J, et al. Immunosuppressive therapy is more effective than interferon in neuromyelitis optica. Mult Scler 2007;13:256-259.
Shimizu Y, Yokoyama K, Misu T, et al. Development of extensive brain lesions following interferon beta therapy in relapsing neuromyelitis optica and longitudinally extensive myelitis. J Neurol 2008;255:305-307.
Cree BA, Lamb S, Morgan K, Chen A, Waubant E, Genain C. An open label study of the effects of rituximab in neuromyelitis optica. Neurology 2005;64:1270-1272.
Lucchinetti CF, Mandler RN, McGavern D, et al. A role for humoral mechanisms in the pathogenesis of Devic's neuromyelitis optica. Brain 2002;125:1450-1461.
Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004;364:2106-2112.
Sellner J, Boggild M, Clanet M, et al. EFNS guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol 2010;17:1019-1032.
Trebst C, Berthele A, Jarius S, et al. [Diagnosis and treatment of neuromyelitis optica. Consensus recommendations of the Neuromyelitis Optica Study Group]. Nervenarzt 2011;82:768-777 (in German).
Kimbrough DJ, Fujihara K, Jacob A, et al. Treatment of neuromyelitis optica: Review and recommendations. Mult Scler Relat Disord 2012;1:180-187.
Palace J, Leite MI, Jacob A. A practical guide to the treatment of neuromyelitis optica. Pract Neurol 2012;12:209-214.
Mitsdoerffer M, Kuchroo V, Korn T. Immunology of neuromyelitis optica: a T cell-B cell collaboration. Ann N Y Acad Sci 2013;1283:57-66..
Papadopoulos MC, Bennett JL, Verkman AS. Treatment of neuromyelitis optica: state-of-the-art and emerging therapies. Nat Rev Neurol 2014;10:493-506.
de Sa JC, Airas L, Bartholome E, et al. Symptomatic therapy in multiple sclerosis: a review for a multimodal approach in clinical practice. Ther Adv Neurol Disord 2011;4:139-168.
Samkoff LM, Goodman AD. Symptomatic management in multiple sclerosis. Neurol Clin 2011;29:449-463.
Abboud H, Petrak A, Mealy M, Sasidharan S, Siddique L, Levy M. Treatment of acute relapses in neuromyelitis optica: Steroids alone versus steroids plus plasma exchange. Mult Scler 2015 Apr 28 [Epub ahead of print].
Kleiter I, Gahlen A, Borisow N, et al. Neuromyelitis optica: Evaluation of 871 attacks and 1153 treatment courses. Ann Neurol 2015 Nov 4 [Epub ahead of print].
Kira J-i, Yamasaki R, Yoshimura S, et al. Efficacy of methylprednisolone pulse therapy for acute relapse in Japanese patients with multiple sclerosis and neuromyelitis optica: A multicenter retrospective analysis – 1. Whole group analysis. Clin Exp Neuroimmunol 2013;4:305-317.
Nakamura M, Nakazawa T, Doi H, et al. Early high-dose intravenous methylprednisolone is effective in preserving retinal nerve fiber layer thickness in patients with neuromyelitis optica. Graefe's Arch Clin Exp Ophthalmol 2010;248:1777-1785.
Weinshenker BG, O'Brien PC, Petterson TM, et al. A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Ann Neurol 1999;46:878-86.
Ruprecht K, Klinker E, Dintelmann T, Rieckmann P, Gold R. Plasma exchange for severe optic neuritis: treatment of 10 patients. Neurology 2004;63:1081-1083.
Keegan M, Konig F, McClelland R, et al. Relation between humoral pathological changes in multiple sclerosis and response to therapeutic plasma exchange. Lancet 2005;36:579-582.
Trebst C, Reising A, Kielstein JT, Hafer C, Stangel M. Plasma exchange therapy in steroid-unresponsive relapses in patients with multiple sclerosis. Blood Purif 2009;28:108-115.
Magana SM, Keegan BM, Weinshenker BG, et al. Beneficial plasma exchange response in central nervous system inflammatory demyelination. Arch Neurol 2011;68:870-878.
Watanabe S, Nakashima I, Misu T, et al. Therapeutic efficacy of plasma exchange in NMO-IgG-positive patients with neuromyelitis optica. Mult Scler 2007;13:128-132.
Bonnan M, Valentino R, Olindo S, Mehdaoui H, Smadja D, Cabre P. Plasma exchange in severe spinal attacks associated with neuromyelitis optica spectrum disorder. Mult Scler 2009;15:487-492.
Merle H, Olindo S, Jeannin S, et al. Treatment of optic neuritis by plasma exchange (add-on) in neuromyelitis optica. Arch Ophthalmol 2012;130:858-862.
Kim SH, Kim W, Huh SY, Lee KY, Jung IJ, Kim HJ. Clinical efficacy of plasmapheresis in patients with neuromyelitis optica spectrum disorder and effects on circulating anti-aquaporin-4 antibody levels. J Clin Neurol 2013;9:36-42.
Lim YM, Pyun SY, Kang BH, Kim J, Kim KK. Factors associated with the effectiveness of plasma exchange for the treatment of NMO-IgG-positive neuromyelitis optica spectrum disorders. Mult Scler 2013;19:1216-1218.
Trebst C, Bronzlik P, Kielstein JT, Schmidt BM, Stangel M. Immunoadsorption therapy for steroid-unresponsive relapses in patients with multiple sclerosis. Blood Purif 2012;33:1-6.
Koziolek MJ, Tampe D, Bahr M, et al. Immunoadsorption therapy in patients with multiple sclerosis with steroid-refractory optical neuritis. J Neuroinflammation 2012;9:80.
Mauch E, Zwanzger J, Hettich R, Fassbender C, Klingel R, Heigl F. [Immunoadsorption for steroid-unresponsive multiple sclerosis-relapses: clinical data of 14 patients]. Nervenarzt 2011;82:1590-1595 (in German).
Koziolek M, Muhlhausen J, Friede T, et al. Therapeutic apheresis in pediatric patients with acute CNS inflammatory demyelinating disease. Blood Purif 2013;36:92-97.
Kobayashi M, Nanri K, Taguchi T, et al. Immunoadsorption therapy for neuromyelitis optica spectrum disorders long after the acute phase. J Clin Apher 2015;30:43-45.
Kieseier BC, Stuve O, Dehmel T, et al. Disease amelioration with tocilizumab in a treatment-resistant patient with neuromyelitis optica: implication for cellular immune responses. JAMA Neurol 2013;70:390-393.
Lauenstein AS, Stettner M, Kieseier BC, Lensch E. Treating neuromyelitis optica with the interleukin-6 receptor antagonist tocilizumab. BMJ Case Rep 2014;2014.
Elsone L, Panicker J, Mutch K, Boggild M, Appleton R, Jacob A. Role of intravenous immunoglobulin in the treatment of acute relapses of neuromyelitis optica: experience in 10 patients. Mult Scler 2014;20:501-504.
Levy M, Mealy MA. Purified human C1-esterase inhibitor is safe in acute relapses of neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm 2014;1:e5.
Saadoun S, Waters P, MacDonald C, et al. Neutrophil protease inhibition reduces neuromyelitis optica-immunoglobulin G-induced damage in mouse brain. Ann Neurol 2012;71:323-333.
Herges K, de Jong BA, Kolkowitz I, et al. Protective effect of an elastase inhibitor in a neuromyelitis optica-like disease driven by a peptide of myelin oligodendroglial glycoprotein. Mult Scler 2012;18:398-408.
Krumbholz M, Hofstadt-van Oy U, Angstwurm K, et al. Very late-onset neuromyelitis optica spectrum disorder beyond the age of 75. J Neurol 2015;262:1379-1384.
Jiao Y, Fryer JP, Lennon VA, et al. Updated estimate of AQP4-IgG serostatus and disability outcome in neuromyelitis optica. Neurology 2013;81:1197-1204.
Shimizu J, Hatanaka Y, Hasegawa M, et al. IFNbeta-1b may severely exacerbate Japanese optic-spinal MS in neuromyelitis optica spectrum. Neurology 2010;75:1423-1427.
Palace J, Leite MI, Nairne A, Vincent A. Interferon beta treatment in neuromyelitis optica: increase in relapses and aquaporin 4 antibody titers. Arch Neurol 2010;67:1016-1017.
Uzawa A, Mori M, Hayakawa S, Masuda S, Kuwabara S. Different responses to interferon beta-1b treatment in patients with neuromyelitis optica and multiple sclerosis. Eur J Neurol 2010;17:672-676.
Kim SH, Kim W, Li XF, Jung IJ, Kim HJ. Does interferon beta treatment exacerbate neuromyelitis optica spectrum disorder? Mult Scler 2012;18:1480-1483.
Barnett MH, Prineas JW, Buckland ME, Parratt JD, Pollard JD. Massive astrocyte destruction in neuromyelitis optica despite natalizumab therapy. Mult Scler 2012;18:108-112.
Kleiter I, Hellwig K, Berthele A, et al. Failure of natalizumab to prevent relapses in neuromyelitis optica. Arch Neurol 2012;69:239-245.
Jacob A, Hutchinson M, Elsone L, et al. Does natalizumab therapy worsen neuromyelitis optica? Neurology 2012;79:1065-1066.
Min JH, Kim BJ, Lee KH. Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Mult Scler 2012;18:113-115.
Kira J, Itoyama Y, Kikuchi S, et al. Fingolimod (FTY720) therapy in Japanese patients with relapsing multiple sclerosis over 12 months: Results of a phase 2 observational extension. BMC Neurol 2014;14:21.
Qian P, Cross AH, Naismith RT. Lack of response to monoclonal antibody therapy in neuromyelitis optica. Arch Neurol 2011;68:1207-1209.
Gelfand JM, Cotter J, Klingman J, Huang EJ, Cree BA. Massive CNS monocytic infiltration at autopsy in an alemtuzumab-treated patient with NMO. Neurol Neuroimmunol Neuroinflamm 2014;1:e34.
Juryńczyk M, Weinshenker B, Akman-Demir G, et al. Status of diagnostic approaches to AQP4-IgG seronegative NMO and NMO/MS overlap syndromes. J Neurol 2015. doi:10.1007/s00415-015-7952-8.
Wingerchuk DM, Weinshenker BG. Neuromyelitis optica. Curr Treat Options Neurol 2008;10:55-66.
Luhder F, Reichardt HM. Traditional concepts and future avenues of glucocorticoid action in experimental autoimmune encephalomyelitis and multiple sclerosis therapy. Crit Rev Immunol 2009;29:255-273.
Watanabe S, Misu T, Miyazawa I, et al. Low-dose corticosteroids reduce relapses in neuromyelitis optica: a retrospective analysis. Mult Scler 2007;13:968-974.
Jarius S, Aboul-Enein F, Waters P, et al. Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain 2008;131:3072-3080.
Bichuetti DB, Lobato de Oliveira EM, Oliveira DM, Amorin de Souza N, Gabbai AA. Neuromyelitis optica treatment: Analysis of 36 patients. Arch Neurol 2010;67:1131-1136.
Sahraian MA, Moinfar Z, Khorramnia S, Ebrahim MM. Relapsing neuromyelitis optica: demographic and clinical features in Iranian patients. Eur J Neurol 2010;17:794-799.
Costanzi C, Matiello M, Lucchinetti CF, et al. Azathioprine: Tolerability, efficacy, and predictors of benefit in neuromyelitis optica. Neurology 2011;77:659-666.
Elsone L, Kitley J, Luppe S, et al. Long-term efficacy, tolerability and retention rate of azathioprine in 103 aquaporin-4 antibody-positive neuromyelitis optica spectrum disorder patients: a multicentre retrospective observational study from the UK. Mult Scler 2014;20:1533-1540.
Mealy MA, Wingerchuk DM, Palace J, Greenberg BM, Levy M. Comparison of relapse and treatment failure rates among patients with neuromyelitis optica: Multicenter study of treatment efficacy. JAMA Neurol 2014;71:324-330.
Qiu W, Kermode AG, Li R, et al. Azathioprine plus corticosteroid treatment in Chinese patients with neuromyelitis optica. J Clin Neurosci 2015;22:1178-1182.
Jeong IH, Park B, Kim SH, Hyun JW, Joo J, Kim HJ. Comparative analysis of treatment outcomes in patients with neuromyelitis optica spectrum disorder using multifaceted endpoints. Mult Scler 2015 Jun 3 [Epub ahead of print]. .
Opelz G, Dohler B. Critical threshold of azathioprine dosage for maintenance immunosuppression in kidney graft recipients. Collaborative Transplant Study. Transplantation 2000;69:818-821.
Jacob A, Matiello M, Weinshenker BG, et al. Treatment of neuromyelitis optica with mycophenolate mofetil: retrospective analysis of 24 patients. Arch Neurol 2009;66:1128-1133.
Huh SY, Kim SH, Hyun JW, et al. Mycophenolate mofetil in the treatment of neuromyelitis optica spectrum disorder. JAMA Neurol 2014;71:1372-1378.
Kitley J, Elsone L, George J, et al. Methotrexate is an alternative to azathioprine in neuromyelitis optica spectrum disorders with aquaporin-4 antibodies. J Neurol Neurosurg Psychiatry 2013;84:918-921.
Ramanathan RS, Malhotra K, Scott T. Treatment of neuromyelitis optica/neuromyelitis optica spectrum disorders with methotrexate. BMC Neurol 2014;14:51.
Minagar A, Sheremata WA. Treatment of Devic’s disease with methotrexate and prednisone. Int J MS Care 2000;2:43-49.
Kageyama T, Komori M, Miyamoto K, et al. Combination of cyclosporine A with corticosteroids is effective for the treatment of neuromyelitis optica. J Neurol 2013;260:627-634.
Tanaka M, Kinoshita M, Tanaka K. Corticosteroid and tacrolimus treatment in neuromyelitis optica related disorders. Mult Scler 2015;21:669.
Zheng X, Zhang X, Liu X, et al. Patient with neuromyelitis optica spectrum disorder combined with Sjogren's syndrome relapse free following tacrolimus treatment. Intern Med 2014;53:2377-2380.
Mok CC, To CH, Mak A, Poon WL. Immunoablative cyclophosphamide for refractory lupus-related neuromyelitis optica. J Rheumatol 2008;35:172-174.
Kim SH, Kim W, Park MS, Sohn EH, Li XF, Kim HJ. Efficacy and safety of mitoxantrone in patients with highly relapsing neuromyelitis optica. Arch Neurol 2011;68:473-479.
Cabre P, Olindo S, Marignier R, et al. Efficacy of mitoxantrone in neuromyelitis optica spectrum: clinical and neuroradiological study. J Neurol Neurosurg Psychiatry 2013;84:511-516.
Weinstock-Guttman B, Ramanathan M, Lincoff N, et al. Study of mitoxantrone for the treatment of recurrent neuromyelitis optica (Devic disease). Arch Neurol 2006;63:957-963.
Yaguchi H, Sakushima K, Takahashi I, et al. Efficacy of intravenous cyclophosphamide therapy for neuromyelitis optica spectrum disorder. Intern Med 2013;52:969-972.
Bichuetti DB, Oliveira EM, Boulos Fde C, Gabbai AA. Lack of response to pulse cyclophosphamide in neuromyelitis optica: evaluation of 7 patients. Arch Neurol 2012;69:938-939.
Torres J, Pruitt A, Balcer L, Galetta S, Markowitz C, Dahodwala N. Analysis of the treatment of neuromyelitis optica. J Neurol Sci 2015;351:31-35.
Miyamoto K, Kusunoki S. Intermittent plasmapheresis prevents recurrence in neuromyelitis optica. Ther Apher Dial 2009;13:505-508.
Kim SH, Kim W, Li XF, Jung IJ, Kim HJ. Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch Neurol 2011;68:1412-1420.
Kim SH, Huh SY, Lee SJ, Joung A, Kim HJ. A 5-year follow-up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol 2013;70:1110-1117.
Pellkofer HL, Krumbholz M, Berthele A, et al. Long-term follow-up of patients with neuromyelitis optica after repeated therapy with rituximab. Neurology 2011;76:1310-1315.
Bedi GS, Brown AD, Delgado SR, Usmani N, Lam BL, Sheremata WA. Impact of rituximab on relapse rate and disability in neuromyelitis optica. Mult Scler 2011;17:1225-1230.
Greenberg BM, Graves D, Remington G, et al. Rituximab dosing and monitoring strategies in neuromyelitis optica patients: creating strategies for therapeutic success. Mult Scler 2012;18:1022-1026.
Kim SH, Jeong IH, Hyun JW, et al. Treatment outcomes with rituximab in 100 patients with neuromyelitis optica: Influence of FCGR3A polymorphisms on the therapeutic response to rituximab. JAMA Neurol 2015;72:989-995.
Yang CS, Yang L, Li T, et al. Responsiveness to reduced dosage of rituximab in Chinese patients with neuromyelitis optica. Neurology 2013;81:710-713.
Ringelstein M, Harmel J, Distelmaier F, et al. Neuromyelitis optica and pregnancy during therapeutic B cell depletion: infant exposure to anti-AQP4 antibody and prevention of rebound relapses with low-dose rituximab postpartum. Mult Scler 2013;19:1544-1547.
Nakashima I, Takahashi T, Cree BA, et al. Transient increases in anti-aquaporin-4 antibody titers following rituximab treatment in neuromyelitis optica, in association with elevated serum BAFF levels. J Clin Neurosci 2011;18:997-998.
Perumal JS, Kister I, Howard J, Herbert J. Disease exacerbation after rituximab induction in neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm 2015;2:e61.
MedImmune. A double-masked, placebo-controlled study with open label period to evaluate MEDI-551 in neuromyelitis optica and neuromyelitis optica spectrum disorders. Available at: https://clinicaltrials.gov/ct2/show/NCT02200770. Accessed May 30, 2015.
Levy M. Ublituximab for acute neuromyelitis optica (NMO) relapses. Available at: https://clinicaltrials.gov/ct2/show/NCT02276963. Accessed May 30, 2015.
van Vollenhoven RF, Emery P, Bingham CO, 3rd, et al. Long-term safety of rituximab in rheumatoid arthritis: 9.5-year follow-up of the global clinical trial programme with a focus on adverse events of interest in RA patients. Ann Rheum Dis 2013;72:1496-1502.
Plate A, Havla J, Kumpfel T. Late-onset neutropenia during long-term rituximab therapy in neuromyelitis optica. Mult Scler Relat Disord 2014;3:269-272.
Kim W, Kim SH, Huh SY, et al. Reduced antibody formation after influenza vaccination in patients with neuromyelitis optica spectrum disorder treated with rituximab. Eur J Neurol 2013;20:975-980.
Rose-John S, Gold R. Devic disease: translational medicine at work. Neurology 2014;82:1294-1295.
Rose-John S. IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int J Biol Sci 2012;8:1237-1247.
Uzawa A, Mori M, Arai K, et al. Cytokine and chemokine profiles in neuromyelitis optica: significance of interleukin-6. Mult Scler 2010;16:1443-1452.
Icoz S, Tuzun E, Kurtuncu M, et al. Enhanced IL-6 production in aquaporin-4 antibody positive neuromyelitis optica patients. Int J Neurosci 2010;120:71-75.
Chihara N, Aranami T, Sato W, et al. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc Natl Acad Sci U S A 2011;108:3701-3706.
Araki M, Aranami T, Matsuoka T, Nakamura M, Miyake S, Yamamura T. Clinical improvement in a patient with neuromyelitis optica following therapy with the anti-IL-6 receptor monoclonal antibody tocilizumab. Mod Rheumatol 2013;23:827-831.
Ayzenberg I, Kleiter I, Schroder A, et al. Interleukin 6 receptor blockade in patients with neuromyelitis optica nonresponsive to anti-CD20 therapy. JAMA Neurol 2013;70:394-397.
Araki M, Matsuoka T, Miyamoto K, et al. Efficacy of the anti-IL-6 receptor antibody tocilizumab in neuromyelitis optica: a pilot study. Neurology 2014;82:1302-1306.
Ringelstein M, Ayzenberg I, Harmel J, et al. Long-term therapy with interleukin 6 receptor blockade in highly active neuromyelitis optica spectrum disorder. JAMA Neurol 2015;72:756-763.
Kanamori Y, Nakashima I, Takai Y, et al. Pain in neuromyelitis optica and its effect on quality of life: a cross-sectional study. Neurology 2011;77:652-658.
Qian P, Lancia S, Alvarez E, Klawiter EC, Cross AH, Naismith RT. Association of neuromyelitis optica with severe and intractable pain. Arch Neurol 2012;69:1482-1487.
Andratsch M, Mair N, Constantin CE, et al. A key role for gp130 expressed on peripheral sensory nerves in pathological pain. J Neurosci 2009;29:13473-13483.
Chugai. Efficacy and safety study as add-on therapy of SA237 to treat NMO and NMOSD. Available at: https://clinicaltrials.gov/ct2/show/NCT02028884. Accessed May 30, 2015.
Chugai. Efficacy and safety study as monotherapy of SA237 to treat NMO and NMOSD. Available at: https://clinicaltrials.gov/ct2/show/NCT02073279. Accessed May 30, 2015.
Hinson SR, Pittock SJ, Lucchinetti CF, et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology 2007;69:2221-2231.
Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain 2010;133:349-361.
Woodruff TM, Nandakumar KS, Tedesco F. Inhibiting the C5-C5a receptor axis. Mol Immunol 2011;48:1631-1642.
Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: Role in immunity. Front Immunol 2015;6:257.
Pittock SJ, Lennon VA, McKeon A, et al. Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol 2013;12:554-562.
Alexion. A double blind trial to evaluate the safety and efficacy of eculizumab in relapsing NMO patients (PREVENT Study). Available at: https://clinicaltrials.gov/ct2/show/NCT01892345. Accessed May 30, 2015.
Khatri BO, Kramer J, Dukic M, Palencia M, Verre W. Maintenance plasma exchange therapy for steroid-refractory neuromyelitis optica. J Clin Apher 2012;27:183-192.
Bakker J, Metz L. Devic's neuromyelitis optica treated with intravenous gamma globulin (IVIG). Can J Neurol Sci 2004;31:265-267.
Okada K, Tsuji S, Tanaka K. Intermittent intravenous immunoglobulin successfully prevents relapses of neuromyelitis optica. Intern Med 2007;46:1671-1672.
Greco R, Bondanza A, Vago L, et al. Allogeneic hematopoietic stem cell transplantation for neuromyelitis optica. Ann Neurol 2014;75:447-453.
Greco R, Bondanza A, Oliveira MC, et al. Autologous hematopoietic stem cell transplantation in neuromyelitis optica: a registry study of the EBMT Autoimmune Diseases Working Party. Mult Scler 2015;21:189-197.
Matiello M, Pittock SJ, Porrata L, Weinshenker BG. Failure of autologous hematopoietic stem cell transplantation to prevent relapse of neuromyelitis optica. Arch Neurol 2011;68:953-955.
Bluestone JA, Bour-Jordan H, Cheng M, Anderson M. T cells in the control of organ-specific autoimmunity. J Clin Invest 2015;125:2250-2260.
Roep BO, Solvason N, Gottlieb PA, et al. Plasmid-encoded proinsulin preserves C-peptide while specifically reducing proinsulin-specific CD8(+) T cells in type 1 diabetes. Sci Transl Med 2013;5:191ra82.
Huang X, Wu H, Lu Q. The mechanisms and applications of T cell vaccination for autoimmune diseases: a comprehensive review. Clin Rev Allergy Immunol 2014;47:219-233.
Lutterotti A, Yousef S, Sputtek A, et al. Antigen-specific tolerance by autologous myelin peptide-coupled cells: a phase 1 trial in multiple sclerosis. Sci Transl Med 2013;5:188ra75.
Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care 2011;34:2026-2032.
Weinshenker BG, Barron G, Behne JM, et al. Challenges and opportunities in designing clinical trials for neuromyelitis optica. Neurology 2015;84:1805-1815.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
IK has received honoraria for consultancy or speaking and travel reimbursement from Bayer Health Care, Biogen Idec, Chugai, and Novartis; RG has received speaker’s and board honoraria from Baxter, Bayer Schering, Biogen Idec, CLB Behring, Genzyme, Merck Serono, Novartis, Talecris, TEVA, and Wyeth. The department of IK and RG has received grant support from Bayer Health Care, Biogen Idec, Genzyme, Merck Serono, Novartis, and TEVA.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1225 kb)
Rights and permissions
About this article

Cite this article
Kleiter, I., Gold, R. Present and Future Therapies in Neuromyelitis Optica Spectrum Disorders. Neurotherapeutics 13, 70–83 (2016). https://doi.org/10.1007/s13311-015-0400-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-015-0400-8