
Abstract
Assessing genetic diversity, population structure, and linkage disequilibrium is important in identifying potential parental lines for breeding programs. In this study, we assessed the genetic and phenotypic variation of 174 normal maize (Zea mays) inbred lines and made association analyses with respect to nine agronomical traits, using 150 simple sequence repeats (SSR). From population structure analysis, the lines were divided into three groups. Association analysis was done with a mixed linear model and a general linear model. Twenty-one marker-trait associations involving 19 SSR markers were observed using the mixed model, with a significance level of P < 0.01. All of these associations, as well as 120 additional marker-trait associations involving 77 SSR markers, were observed with the general model. Two significant marker-trait associations (SMTAs) were detected at P ≤ 0.0001. In the mixed linear model, one locus was associated with water content, two loci were associated with 100-kernel weight, setted ear length, ear thickness and stem thickness; three loci were associated with ear height, four loci were associated with total kernel weight and five loci were associated with plant height. These results should prove useful to breeders in the selection of parental lines and markers.
Similar content being viewed by others

Introduction
As the world’s population continues to increase, it is imperative that the scientific community use all avenues available to enable growers to meet its ever-increasing need for food and other resources. Modern breeding technologies hold promise in this quest. Maize (Zea mays), with its high photosynthetic rates and yields, is a particularly valuable agricultural resource (Xu et al. 2009), serving as a food source for humans and livestock as well as a biofuel and source of fiber in some parts of the world. Phenotypic diversity for yield-related traits, such as plant height, date of maturity, flowering time, and ear and kernel traits exists in maize, as well as diversity in its response to variable environmental conditions. Growers and breeders have, for many years, exploited this diversity to generate varieties with particular traits (Yan et al. 2011).
Modern breeding technologies hold promise for furthering these endeavors. Inherent in any breeding program is basic knowledge about genetic diversity and population structure in the breeding material (Van Inghelandt et al. 2010). This knowledge permits prediction of the degree of inheritance, variation, and level of heterosis (Qi-Lun et al. 2008). Genetic diversity and population structure can be estimated with molecular markers. Unlike morphological markers, they are not influenced by environmental factors and are usually inherited by Mendelian rules (Legesse et al. 2007). Microsatellite and simple sequence repeats (SSR) are the preferred molecular markers for assessing genetic diversity, as they are codominant, reproducible, tend to show high polymorphism, and broadly distributed throughout the genome (Akagi et al. 1997). SSR are short regions with simple repeated motifs of one to six nucleotide fragments and occur in both coding and non-coding region (Miah et al. 2013).
The association between phenotypic and molecular diversity revealed molecular markers can generate useful information regarding population structure and genetic diversity. Association mapping exploits historical and evolutionary recombination at the population level. Association mapping, which is based on linkage disequilibrium (LD) studies statistical association between genetic markers and phenotype is widely used to identify quantitative trait loci (QTL) in crops (Wei et al. 2014), partly because it has higher mapping resolution than linkage mapping. Other advantages are that many alleles can be evaluated simultaneously and it is possible to assess many recombination events, in numerous generations, and in natural populations without developing a mapping population (Yu and Buckler 2006; Abdurakhmonov and Abdukarimov 2008; Gupta et al. 2014). Association mapping of yield-related traits has been applied to soybean (Hu et al. 2014), rice (Agrama et al. 2007; Borba et al. 2010), rapeseed (Cai et al. 2014), and maize (Xue et al. 2013).
A collection of maize inbred lines of varying origins has been assembled at the Maize Experiment Station operated by the Gangwon Agricultural Research and Extension Services, Hongcheon, Republic of Korea. Most of the lines are appropriate and available for use as parental lines in a breeding program. Our objective in this study was to estimate genetic diversity and population structure of 174 normal maize inbred lines. We performed association mapping of 150 SSR primer sets and nine agronomic traits to evaluate genetic diversity between the lines and the statistical association between genetic markers and phenotype traits.
Materials and methods
Plant material and phenotypic analysis
A total of 174 maize inbred lines were obtained from the Maize Experiment Station of the Gangwon Agricultural Research and Extension Service collected from Korea. Details of the lines are listed in Table 1. A total of nine agronomic traits were evaluated in 2013 and 2014: plant height, ear height, stem thickness, 100-kernel weight, water content, ear length, setted ear length, total kernel weight, and ear thickness.
DNA extraction and SSR amplification
Genomic DNA was extracted from the young leaves by the method of Dellaporta et al. (1983), with minor modifications. One-hundred fifty SSR primer sets derived from Maize GDB (http://www.maizegdb.org/) distributed across the 10 maize chromosomes (2n = 20) were used for analysis of genetic diversity, population structure, and association mapping.
SSR amplifications were performed in a total volume of 30 μL and consisted of 20 ng genomic DNA, 1 × PCR buffer, 0.3 μM forward and reverse primers, 0.2 mM dNTPs, and 1 unit of Taq Polymerase (Bio Tools, Jupiter, FL). The PCR procedure consisted of an initial denaturation of 94 °C for 5 min, followed by two denaturating cycles at 94 °C for 1 min, annealing at 65 °C for 1 min, and extension at 72 °C for 2 min. After the second cycle, the annealing temperature was decreased by 1 °C in every second cycle until it reached 55 °C. The last cycle was repeated 20 times. When the cycles were completed, the extension cycle was extended for 10 min at 72 °C.
Electrophoresis and fragment detection
The SSR reaction products were added to an equal volume of stop solution (98 % deionized formamide, 2 mM EDTA, 0.05 % bromophenol blue, 0.05 % xylene cyanol) and heated at 95 °C for 5 min. A 3-μL aliquot of each reaction mixture was analyzed by 6 % denaturing polyacrylamide gel. Electrophoresis was conducted at 1800 V for 90 min. The separated fragments were then visualized by silver staining.
Data analysis
Phenotype
Pearson correlation coefficients, means, and standard deviation for nine agronomic traits were calculated with an SPSS tool. We have combined the data from both years of the study.
Genotype
The number of alleles, allele frequency, major allele frequency, gene diversity, and polymorphic information content were estimated using powerMarker version 3.25 (Liu and Muse 2005). Genetic similarities were calculated for each pair of lines with the Dice similarity index (Dice 1945). The similarity matrix was then used to construct a dendrogram based on an unweighted-pair-group-methods-using-arithmetic-averages algorithm (UPGMA), with the help of SAHN-Clustering from NTSYS-pc.V.2.1 (Rohlf 1998).
Population structure
The model-based program STRUCTURE 2.2 (Pritchard and Wen 2003) was used to evaluate the population structure. The STRUCTURE program was run five times for each K value, ranging from 1 to 10, using the admixture model with burn-in of 100,000 and run length of 100,000. An average likelihood value [LnP(D)], across all runs was calculated for each K. The ad hoc criterion (ΔK) of Evanno et al. (2005) was used to determine the most probable K value, in order to compensate for overestimation of subgroup number by STRUCTURE. The run of estimated numbers of subgroups showing maximum likelihood was used to assign inbred lines that had membership probabilities of ≥0.80 to subgroups. Inbred lines with membership probabilities of <0.80 were assigned to the admixed group (Stich et al. 2005).
LD and association mapping
Association mapping was performed for marker-trait association using TASSEL 3.0 (Bradbury et al. 2007). We used to models to confirm marker-trait association. These were the general linear model (Q GLM) and mixed linear model (Q + K MLM). The Q GLM method was performed using the population structure (Q) matrix derived from the STRUCTURE program. The number of permutation run was set as 10,000 to obtain a P value for marker significance of 0.05 and 0.01. The Q + K MLM method used a kinship (K) matrix and population structure Q matrix at P < 0.05 and P < 0.01. The K matrix was created in SPAGeDi software (Hardy and Vekemans 2002) by calculating the kinship coefficient using the method of Loiselle et al. (1995).
Results
Phenotypic analysis and correlation analysis
Phenotypic variations of nine agronomic traits in 174 inbred lines were evaluated. Table 2 shows the results of correlation analyses. The strongest correlations, along with the correlation coefficients, were identified as those between: plant height and ear height (0.749**) and ear length and setted ear length (0.756**). Correlation coefficient between total kernel weight and setted ear length (0.514**) was on higher side compared to other significant relationship. P values for these correlations ranged from 0.01 to 0.05 (Table 2).
Genetic variation and diversity among normal maize inbred lines
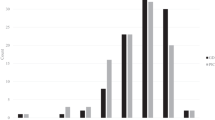
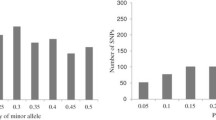
Genetic diversity was evaluated by screening the lines with the SSR loci, which were randomly distributed across the genome. All 150 SSR loci were polymorphic across the inbred lines and a total of 1082 alleles were detected (Supplementary Table 1). The number of alleles per locus ranged from 2 to 20 with an average of 7.21 alleles per locus (Fig. 1a). The average polymorphic information content value was 0.60, with a range of 0.19–0.91. Genetic diversity ranged from 0.20 to 0.92, with an average of 0.65. The average major allele frequency was 0.48, with a range of 0.14–0.89 (Fig. 1b; Table 3). Analysis of all alleles led to identifying 46.11 % (499/1082) as rare (frequency < 0.05), of which 20.14 % (218) were private alleles, only detected in one of the 174 lines. We identified 48.15 % (521 alleles) as intermediate (frequency 0.05–0.5) and 5.73 % (62 alleles) as abundant (frequency > 0.5; Fig. 2).

Frequency of allele number, gene diversity, and polymorphic information content per locus in waxy and normal maize inbred lines

Histogram of allele frequencies in 174 normal maize inbred lines
Population structure and cluster analysis
To investigate genetic relationship among the inbred lines, a model-based program, STRUCTURE, was used to subdivide the inbred lines into appropriate subgroups. It proved difficult to choose K values calculated from five replicate sets ranging from 1 to 10 from the natural logarithm of probability of the data, so we applied the ad hoc measure ΔK, as has been suggested by Evanno et al. (2005). For all lines, the greatest ΔK value was apparent when K = 2. Based on the membership probability threshold of 0.8 (Wang et al. 2008), inbred lines were divided into three groups: I, II, and an admixed group. Thirty-two lines were assigned to group I, 75 to group II, and 67 to the admixed group; the membership threshold was <0.8 (Fig. 3). A dendrogram using UPGMA analysis showed that the clustering pattern was complex. If the genetic similarity value was 35 %, eight groups of clusters were observed. Group I accounted for most, or 113 accessions; group II contained 7 accessions; group III contained 29 accessions; group IV contained 13 accessions; group V contained 8 accessions; group VI contained 2 accessions; and groups VII and VIII each contained one accession (Fig. 3).

Unweighted-pair-group-methods-using-arithmetic-averages algorithm dendrogram and population structure based on 150 simple sequence repeat markers
LD and association mapping using QGLM and Q + K MLM models
LD among SSR locus was investigated in the inbred lines. The LD pattern was assessed based on combinations of the 150 SSR loci. A significant LD value of 20.95 % was observed (P < 0.01) and D’ values that ranged from 0.136 to 0.189, with an average of 0.148 (Table 4). Using a Q GLM model, we detected 120 marker-trait associations involving 77 SSR markers that were associated with nine agronomic traits (Supplementary Table 2). With a Q + K MLM model, there were 21 marker-trait associations involving 19 markers (Supplementary Table 3). Table 5 presents the information on overlapping significant marker trait association (SMTA) between Q GLM and Q + MLM at a significant level of P ≤ 0.01. Among the 21 marker-trait associations, one locus was associated with water content, two loci were associated with 100-kernel weight, setted ear length, ear thickness and stem thickness; three loci were associated with ear height, four loci were associated with total kernel weight and five loci were associated with plant height. Two loci showed association for two traits: umc1581 was associated with both ear height and plant height, umc1904 with both total kernel weight and water content. Two loci, umc1997 and umc2352 associated with 100 kernel weight and Ear thickness were detected at P ≤ 0.0001 in both the models Q GLM and Q + K MLM respectively.
Discussion
Association mapping with molecular markers is a powerful tool for characterizing genetic diversity and population structure (Yan et al. 2011). SSR markers are ideal in this endeavor, due to their abundance, co-dominance, and locus specificity (Gupta et al. 2014). A large germplasm collection is also of value in evaluating population structure and genetic diversity (Gupta et al. 2014) as it permits the selection of diverse plants, an important step for any marker-assisted selection or breeding program (Van Inghelandt et al. 2010). These considerations influenced our experiments. We used association mapping to evaluate genetic diversity and population structure in 174 inbred lines, using 150 SSR primer sets that were distributed across the maize genome. Many other association mapping studies, with various sample sizes, have been conducted in maize inbred lines (Wang et al. 2008; Xie et al. 2008; Van Inghelandt et al. 2010; Wang et al. 2011). The allele frequencies, polymorphic information content values, and gene diversity values that we observed lead us to conclude that the inbred lines contain extensive genetic variation and are a valuable resource for association studies.
Population structure in our data was investigated using a model-based clustering method (STRUCTURE) and distance-based phylogenetic methods (NTSYs). Information on subgrouping of germplasm is vital for hybrid breeding in maize and has important evolutionary and conservation implications. In our analysis, the lines could be divided into two distinct groups (I & II) and a third admixed group. A small percentage of the lines were in group I (18 %) and the other two groups contained the rest, in approximately equally numbers. The lines in admixed group are those that have ancestry from both subpopulations. Custer analysis yielded more information on genetic diversity of the lines. Our analysis was based on UPGMA aided by SAHN-Clustering that was, in turn, based on genetic distance. A complex dendrogram with six major clusters and many subgroup clusters was observed (Fig. 3). The natural history of the pre-domesticated ancestral populations as well as domestication (breeding) practices influence population structure (Xie et al. 2008). The complex evolutionary and breeding history in maize makes population structure, clustering patterns, and genetic background complex. The information from our analysis should enhance understanding of the structural organization of unknown lines of maize, even in the absence of a pedigree and source information. Additionally, the information about population structure that we have identified should be useful in selection of potentially successful crosses between these lines.
The advantages of association makes the former a promising tool for breeders (Yu and Buckler 2006), particularly as compared to linkage mapping. The level of LD among the marker alleles and alleles affecting the phenotype determines the success of association studies (Xie et al. 2008; Yan et al. 2011), and the levels of LD in this study allowed us to detect marker-phenotype associations in the inbred lines. Population structure analysis can lead to many false positive results and a variety of methods, specifically, the principle component model, the Q + K model, genomic control, and structured association, are used to minimize errors (Yu and Buckler 2006). A combined model is used for mixed model frameworks (Q + K) for marker-trait associations (Yu et al. 2006; Zhao et al. 2007; Stich and Melchinger 2009). Typically, data obtained from random markers to estimate the population structure matrix (Q) and the relative kinship matrix (K) is used for mixed model framework (Q + K) for marker-trait associations (Yu et al. 2006; Zhao et al. 2007; Stich and Melchinger 2009). Here, we used a general linear model based on the Q matrix (Q GLM) and a mixed linear model based on the Q and K matrices (Q + K MLM) for analysis of data that had been combined from two consecutive years (Supplementary Tables 2 and 3).
Based on the Q GLM model, 120 marker-trait associations were identified for nine agronomic traits involving 77 markers. Based on the Q + K MLM model, 21 marker-trait associations were identified involving 19 markers. All the statistically significant associations (P < 0.01) that were detected with the MLM approach were supported by the GLM approach. Based on significant marker trait association two markers umc1997 and umc2352 associated with 100 kernel weight and Ear thickness were detected at P ≤ 0.0001. We also observed two markers associated with two traits. Specifically, umc1581 located on chromosome 2 associated with ear height and plant height; umc1904, located on chromosome 8, were associated with total kernel weight and water content. The locus located on chromosome region 4, umc1652, which associated with plant height in other association analyses (Zhang et al. 2012), was also associated with plant height in our study.
Many other association mapping studies, with various sample sizes, have been conducted in maize inbred lines (Wang et al. 2008; Xie et al. 2008; Van Inghelandt et al. 2010; Wang et al. 2011). Zhang et al. 2012 concluded that umc1652 and umc1695 were associated with plant height and cob diameter, respectively. Our results confirm that umc1652 associates with plant height, but we observed that umc1695 associated with stem thickness. QTLs for grain weight (Zheng et al. 2009) involving umc1166 was associated with stem thickness, in our study.
In conclusion, appropriate parental selection is an important and basic step for any successful breeding program, especially if the focus is on yield-related traits. Association analysis aids the assessment of genetic diversity, population structure, LD, and marker-trait association. This information should prove useful to breeding programs by informing the choice of appropriate lines and identifying loci that affect agronomical traits. The information from our study regarding loci associated with yield and agronomic traits may permit marker-assisted selection.
References
Abdurakhmonov IY, Abdukarimov A (2008) Application of association mapping to understanding the genetic diversity of plant germplasm resources. Int J Plant Genomics 2008:574927
Agrama HA, Eizenga GC, Yan W (2007) Association mapping of yield and its components in rice cultivars. Mol Breed 19:341–356
Akagi H, Yokozeki Y, Inagaki A, Fujimura T (1997) Highly polymorphic microsatellites of rice consist of AT repeats, and a classification of closely related cultivars with these microsatellite loci. Theor Appl Genet 94:61–67
Borba TCO, Brondani RPV, Breseghello F, Coelho ASG, Mendonça JA, Rangel PHN, Brondani C (2010) Association mapping for yield and grain quality traits in rice (Oryza sativa L.). Genet Mol Biol 33:515–524
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Cai D, Xiao Y, Yang W, Ye W, Wang B, Younas M, Wu J, Liu K (2014) Association mapping of six yield-related traits in rapeseed (Brassica napus L.). Theor Appl Genet 127:85–96
Dellaporta SL, Wood J, Hicks JB (1983) A plant DNA minipreparation: version II. Plant Mol Biol Rep 1:19–21
Dice LR (1945) Measures of the amount of ecologic association between species. Ecology 26:297–302
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Gupta S, Kumari K, Muthamilarasan M, Parida SK, Prasad M (2014) Population structure and association mapping of yield contributing agronomic traits in foxtail millet. Plant Cell Rep 33:881–893
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyze spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Hu Z, Zhang D, Zhang G, Kan G, Hong D, Yu D (2014) Association mapping of yield-related traits and SSR markers in wild soybean (Glycine soja Sieb. and Zucc.). Breed Sci 63(5):441–449
Legesse BW, Myburg AA, Pixley KV, Botha AM (2007) Genetic diversity of African maize inbred lines revealed by SSR markers. Hereditas 144:10–17
Liu K, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
Loiselle BA, Sork VL, Nason J, Graham C (1995) Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am J Bot 82:1420–1425
Miah G, Rafii MY, Ismail MR, Puteh AB, Rahim HA, Islam KhN, Latif MA (2013) A review of microsatellite markers and their applications in rice breeding programs to improve blast disease resistance. Int J Mol Sci 14:22499–22528
Pritchard, J. K and Wen. W (2003) Documentation for STRUCTURE software: Version 2
Qi-Lun Y, Ping F, Ke-Cheng K, Guang-Tang P (2008) Genetic diversity based on SSR markers in maize (Zea mays L.) landraces from Wuling mountain region in China. J Genet 87:287–291
Rohlf FJ (1998) NTSYS-pc: Numerical taxonomy and multivariate analysis system. Version: 2.02. Exeter Software, Setauket
Stich B, Melchinger AE (2009) Comparison of mixed-model approaches for association mapping in rapeseed, potato, sugar beet, maize, and Arabidopsis. BMC Genomics 10:94
Stich B, Melchinger AE, Frisch M, Maurer HP, Heckenberger M, Reif JC (2005) Linkage disequilibrium in European elite maize germplasm investigated with SSRs. Theor Appl Genet 111:723–730
Van Inghelandt D, Melchinger AE, Lebreton C, Stich B (2010) Population structure and genetic diversity in a commercial maize breeding program assessed with SSR and SNP markers. Theor Appl Genet 120:1289–1299
Wang R, Yu Y, Zhao J, Shi Y, Song Y, Wang T, Li Y (2008) Population structure and linkage disequilibrium of a mini core set of maize inbred lines in China. Theor Appl Genet 117:1141–1153
Wang M, Zhang X, Zhao J, Song W, Zheng Y (2011) Evaluation of the genetic diversity and genome-wide linkage disequilibrium of Chinese maize inbred lines. Aust J Crop Sci 5:1790–1795
Wei Z, Zhang G, Du Q, Zhang J, Li B, Zhang D (2014) Association mapping for morphological and physiological traits in Populus simonii. BMC Genet 15(Suppl 1):S3
Xie CX, Warburton M, Li MS, Li XH, Xiao MJ, Hao ZF, Zhao Q, Zhang SH (2008) An analysis of population structure and linkage disequilibrium using multilocus data in 187 maize inbred lines. Mol Breed 21:407–418
Xu Y, Skinner DJ, Wu H, Palacios-Rojas N, Araus JL, Yan J, Gao S, Warburton ML, Crouch JH (2009) Advances in maize genomics and their value for enhancing genetic gains from breeding. Int J Plant Genomics 2009:957602
Xue Y, Warburton ML, Sawkins M, Zhang X, Setter T, Xu Y, Grudloyma P, Gethi J, Ribaut J-M, Li W, Zhang X, Zheng Y, Yan J (2013) Genome-wide association analysis for nine agronomic traits in maize under well-watered and water-stressed conditions. Theor Appl Genet 126:2587–2596
Yan J, Warburton M, Crouch J (2011) Association mapping for enhancing maize (Zea mays L.) genetic improvement. Crop Sci 51:433–449
Yu J, Buckler ES (2006) Genetic association mapping and genome organization of maize. Curr Opin Biotech 17:155–160
Yu J, Pressoir G, Briggs WH, Bi IV, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovich S, Buckler ES (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Zhang Q, Wu C, Ren F, Li Y, Zhang C (2012) Association analysis of important agronomical traits of maize inbred lines with SSRs. Aust J Crop Sci 6:1131–1138
Zhao K, Aranzana MJ, Kim S, Lister C, Shindo C, Tang C, Toomajian C, Zheng H, Dean C, Marjoram P, Nordborg M (2007) An Arabidopsis example of association mapping in structured samples. PLoS Genet 3:71–82
Zheng HJ, Wu AZ, Zheng CC, Wang YF, Cai R, Shen XF, Xu RR, Liu P, Kong LJ, Dong ST (2009) QTL mapping of maize (Zea mays) stay-green traits and their relationship to yield. Plant Breed 128:54–62
Acknowledgments
This study was supported by a Korea Research Foundation Grant funded by the Next-Generation BioGreen 21 Program (Plant Molecular Breeding Center, No. PJ0080182014) of the Rural Development Administration, Republic of Korea, and Golden Seed Project (No. 213001-04-1-SBA10), Ministry of Agriculture, Food and Rural Affairs (MAFRA), Ministry of Oceans and Fisheries (MOF), Rural Development of Korea (RDA), and Korea Forest Service (KFS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Additional information
Jong Yeol Park, R. V. Ramekar, and K. J. Sa contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Park, J.Y., Ramekar, R.V., Sa, K.J. et al. Genetic diversity, population structure, and association mapping of biomass traits in maize with simple sequence repeat markers. Genes Genom 37, 725–735 (2015). https://doi.org/10.1007/s13258-015-0309-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-015-0309-y